
What are pyridines?
Pyridine is a colorless liquid that boils at 115 ºC. It used to be obtained almost exclusively from coal tar. Today, it is obtained industrially from ammonia and acetylene.
Pyridine like benzene has 6 π electrons (aromatic Hückel). Its structure is planar with bond angles of 120º. The heat of combustion reveals that the resonance energy is 23 kcal·mol-1.
Like benzene, it has the aromatic sextet, but unlike 5-membered heterocycles, the nitrogen electron pair is not involved in aromaticity. Pyridine exhibits basic properties. The introduction of a heteroatom into the benzene ring makes more canonical structures possible in the resonance hybrid. Specifically, five resonance structures for the case of pyridine.
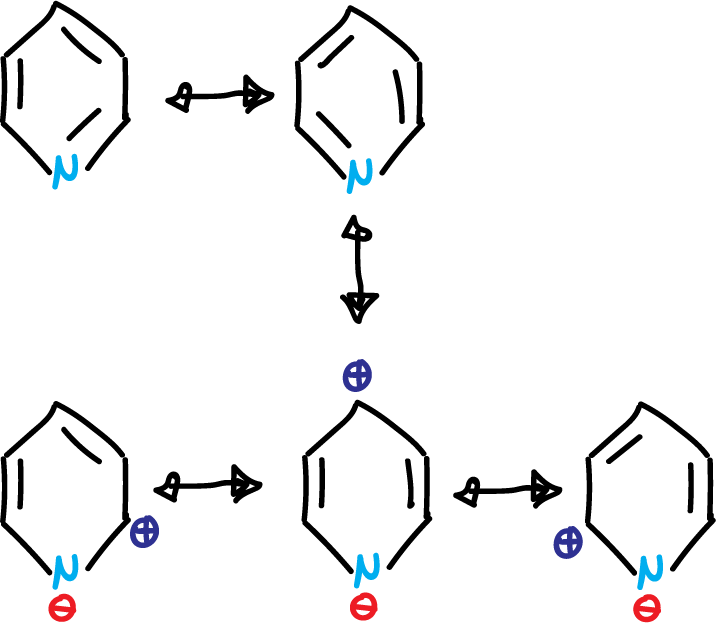
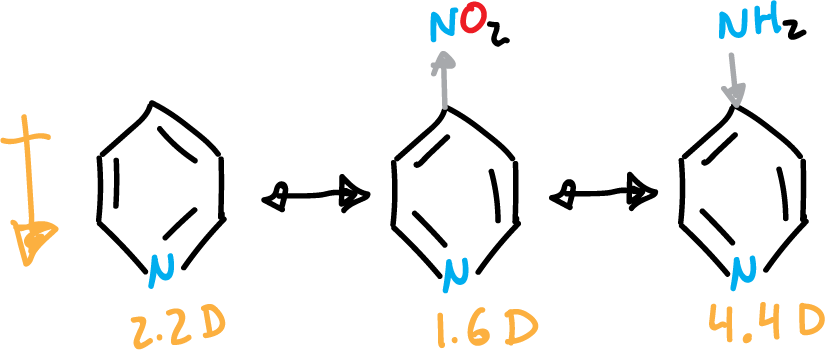
Due to the last 3 highlighted in the figure, the ring is left with a positive charge remainder (⊕), which hinders electrophilic aromatic substitution (SEAr). Also, it makes the pyridine present dipole moment (μ = 2.2 Debye). The existence of an electron-donating group in the C4 position increases this dipole moment and an electron-attracting group in the same position decreases it.
By having a nitrogen atom with an electron pair that is not involved in aromaticity. Consequently, pyridines can be protonated to form the corresponding conjugated acids, or pyridinium ions.
The base strength of pyridines varies according to the nature of the ring substituent. Thus, electron-donating groups increase the pKa value, while electron-attracting groups decrease it.
Thus, pyridines can form complexes with a wide variety of Lewis acids. That is, they react with alkylating agents to give pyridinium salts. They can also be oxidized to give pyridine N-oxides by treatment with peracids.
The pyridine N-oxides, from the structural point of view, are relevant compounds since they have the ability to increase the electron density in various positions of the ring. Or vice versa, depending on the reagent in front of which they are found.
This phenomenon is evident in the structures contributing to the pyridine N-oxide resonance hybrid. As in the case of pyridine, the dipolar forms contribute considerably.
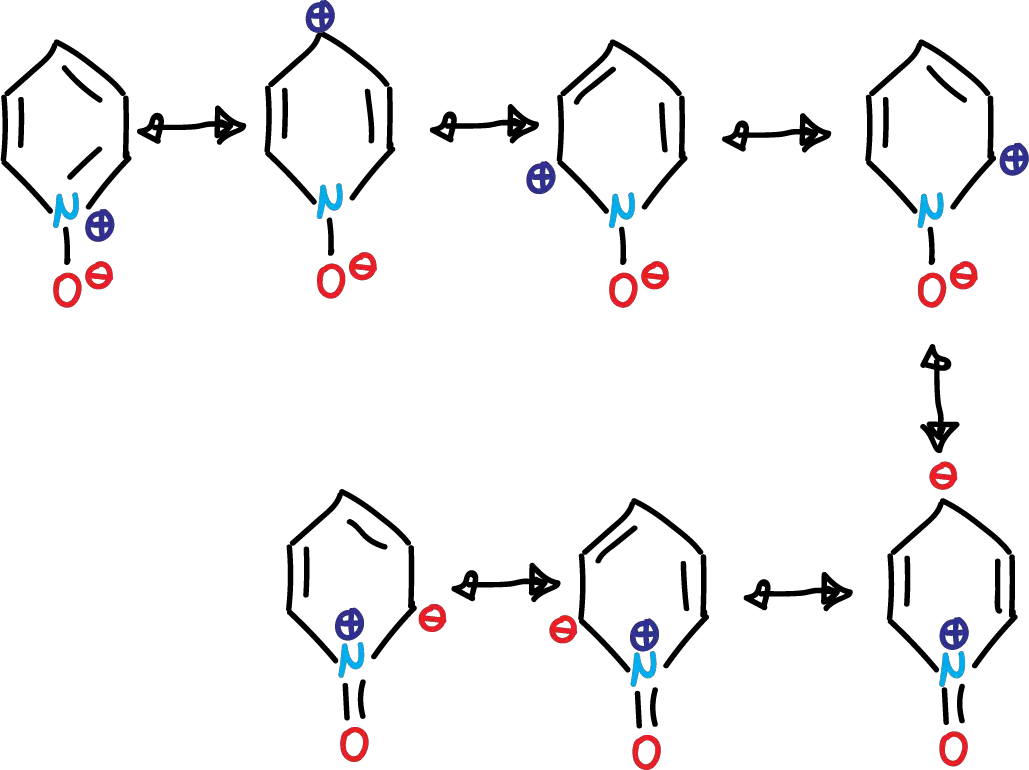
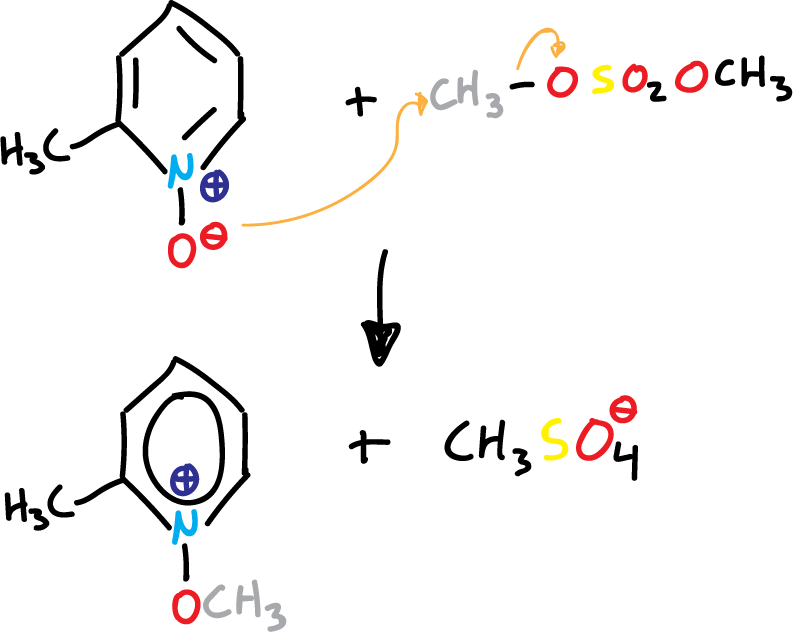
If we look at the first 4 structures, the oxygen atom must be able to act as a nucleophile. This is evident in the reactions of pyridine N-oxides with a variety of alkylating agents. Thus, the result is an SN2-type shift to form N-alkoxypyridinium salts.
Synthesis of pyridines
The classical methods to obtain pyridines from acyclic precursors use the cyclization reactions.
Thus, taking into account a retrosynthetic analysis:
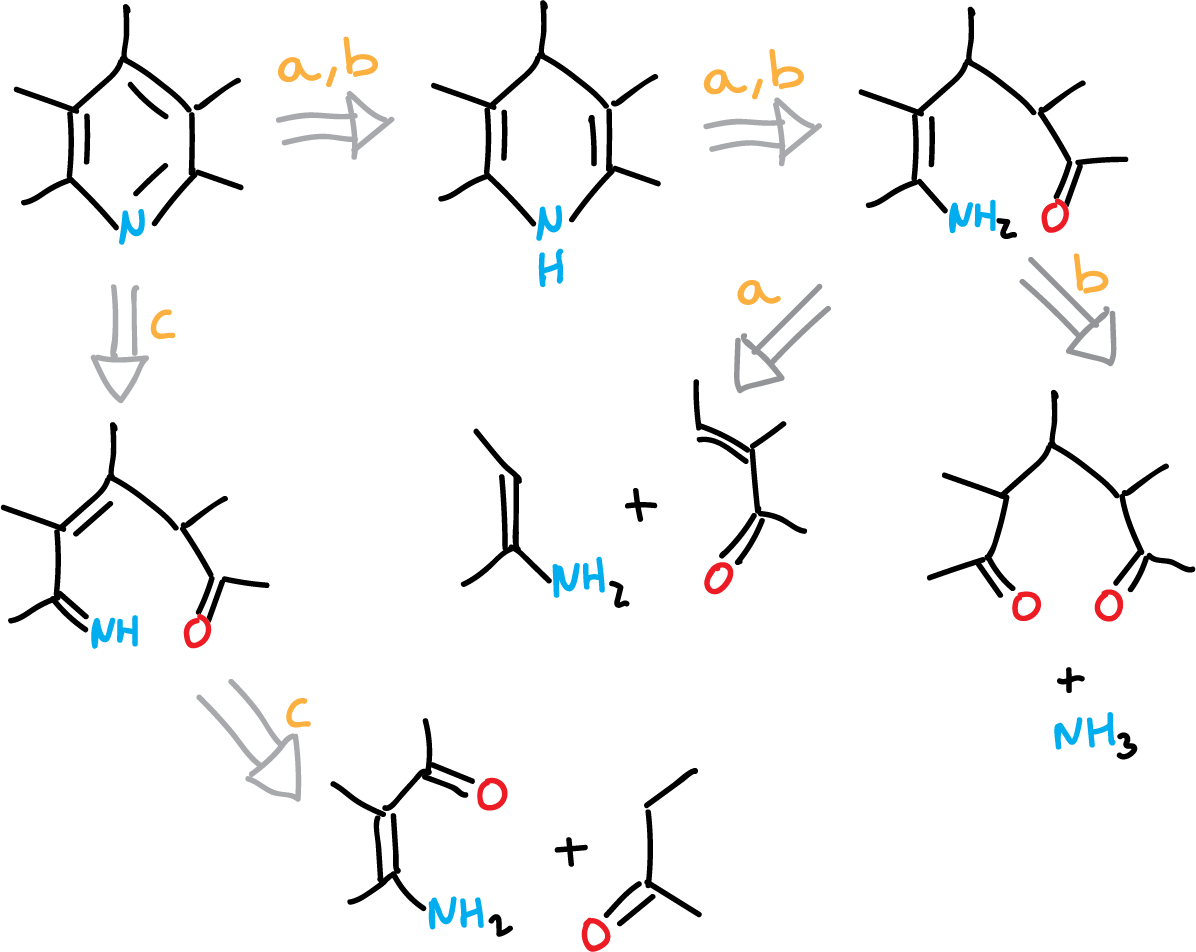
We would have three ways to form the ring from readily available raw materials.
These three methods (routes a, b and c) along with additional ones are used for the synthesis of pyridines.
Hantzsch pyridine synthesis
It consists of the condensation of 2 molecules of a β-ketoester with an aldehyde and ammonia (route a).
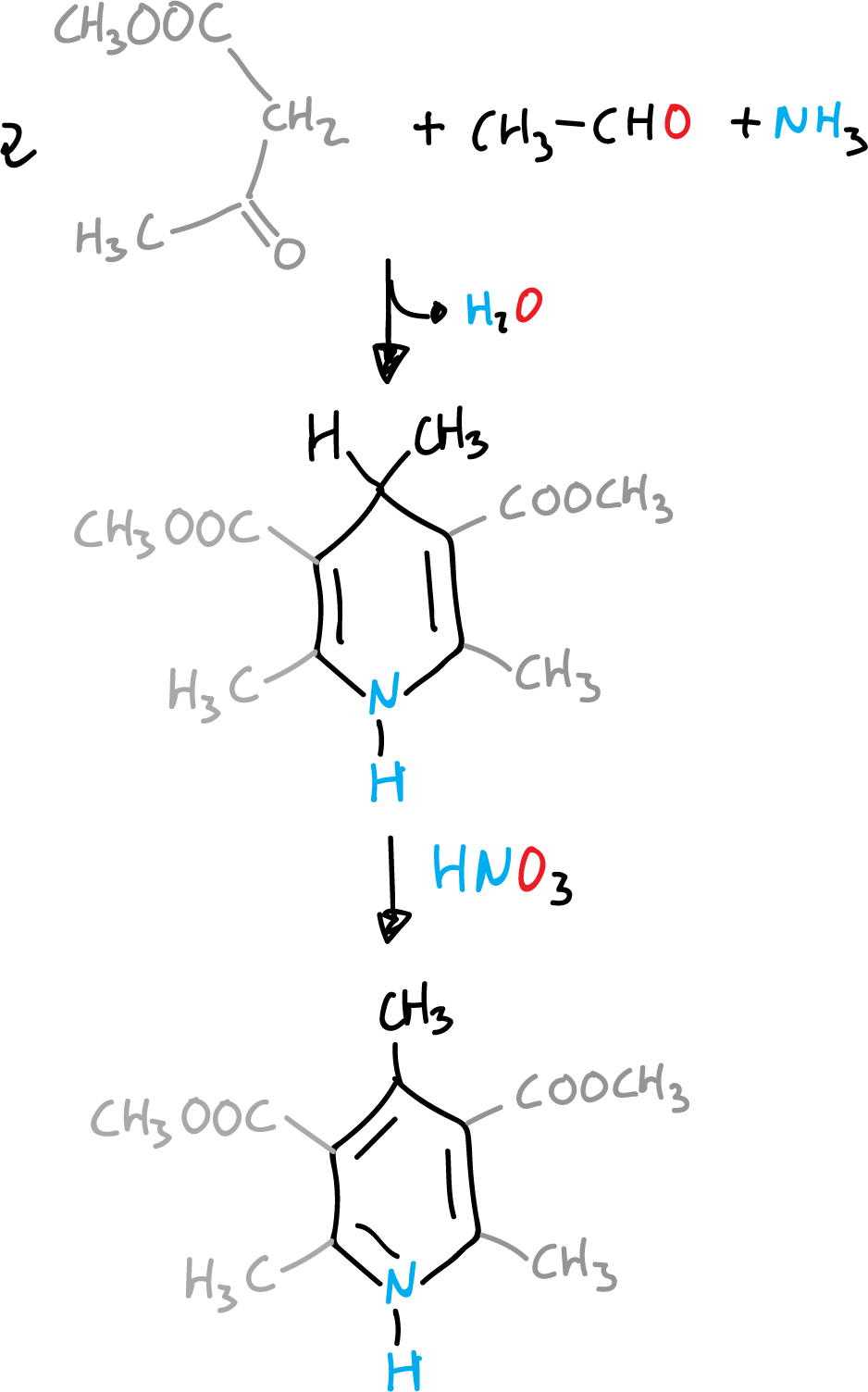
Reaction mechanism
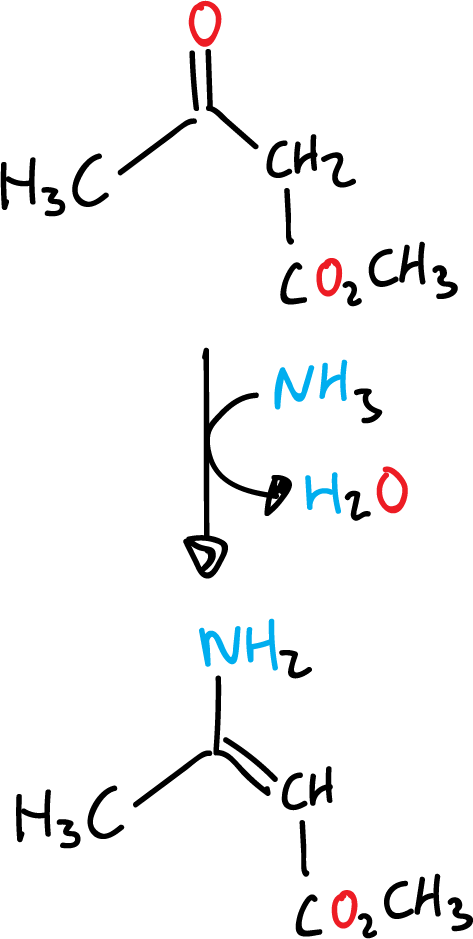
The first stage is a Knoevenagel condensation the β-ketoester and the aldehyde.
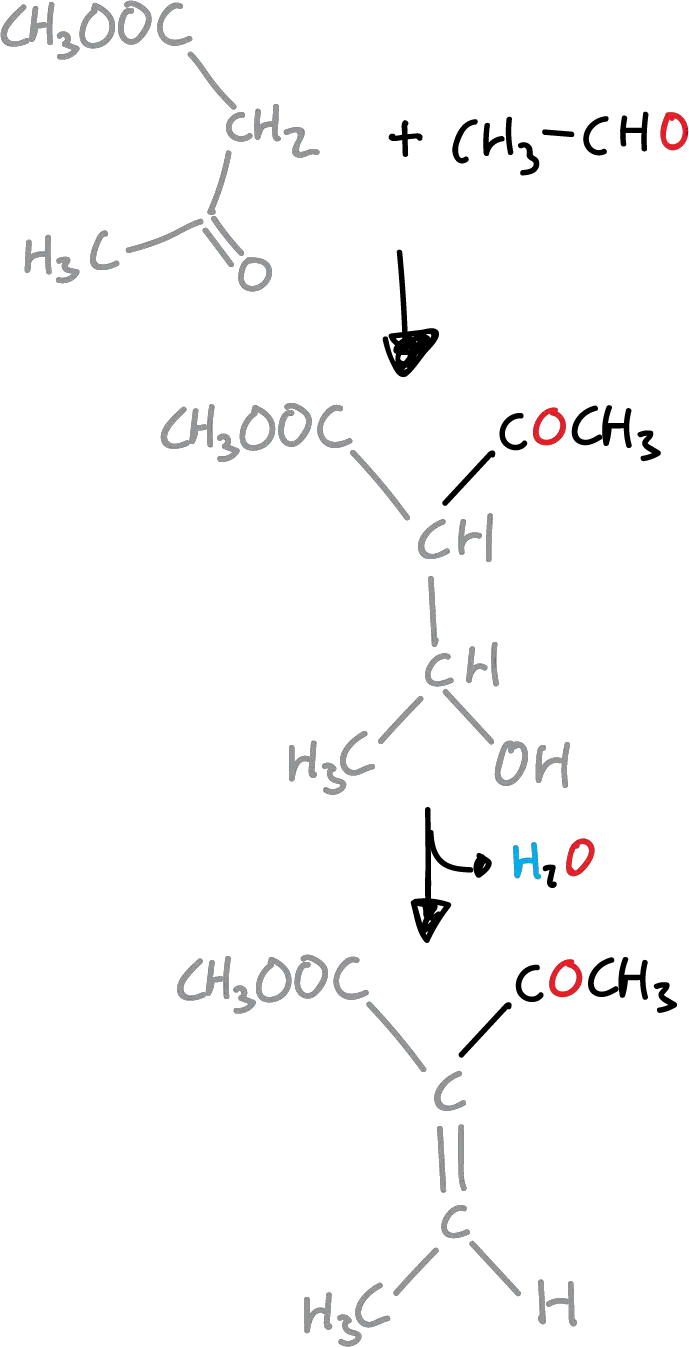
Next, the unsaturated ketoester formed undergoes condensation with the enamine produced with ammonia and the original β-ketoester to give the corresponding dihydropyridine.
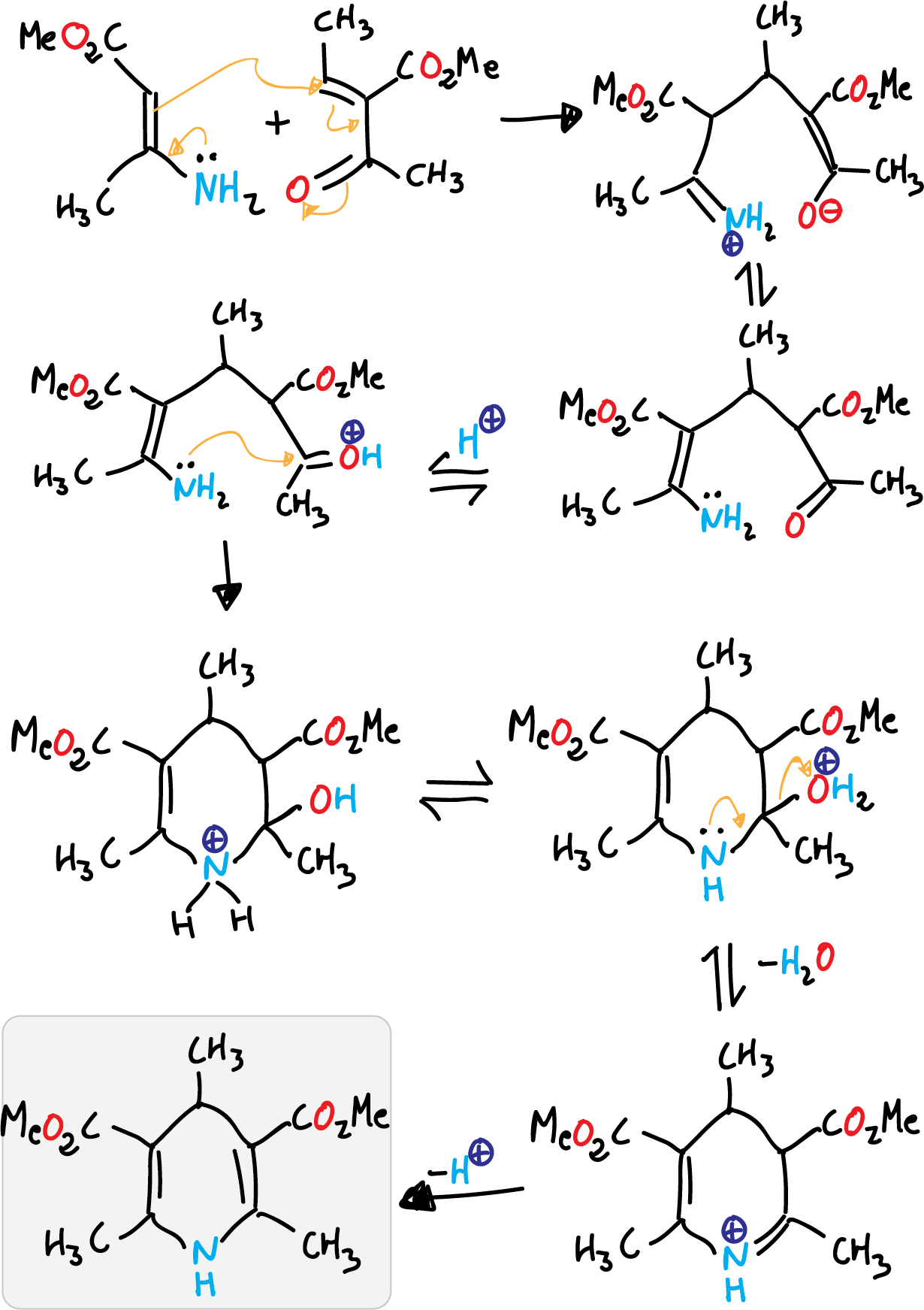
This reaction corresponds to route A of the retrosynthetic analysis previously performed.
Other variants
Many modifications and variants of the Hantzsch synthesis of pyridines have been described.
For example, ammonium acetate (AcONH4) with acetic acid (AcOH) can be used instead of ammonia. This method follows route c of the previous retrosynthetic analysis. Thus, in the first step enamine is formed, which in the following figure is indicated as I reacting with another molecule of the reagent.
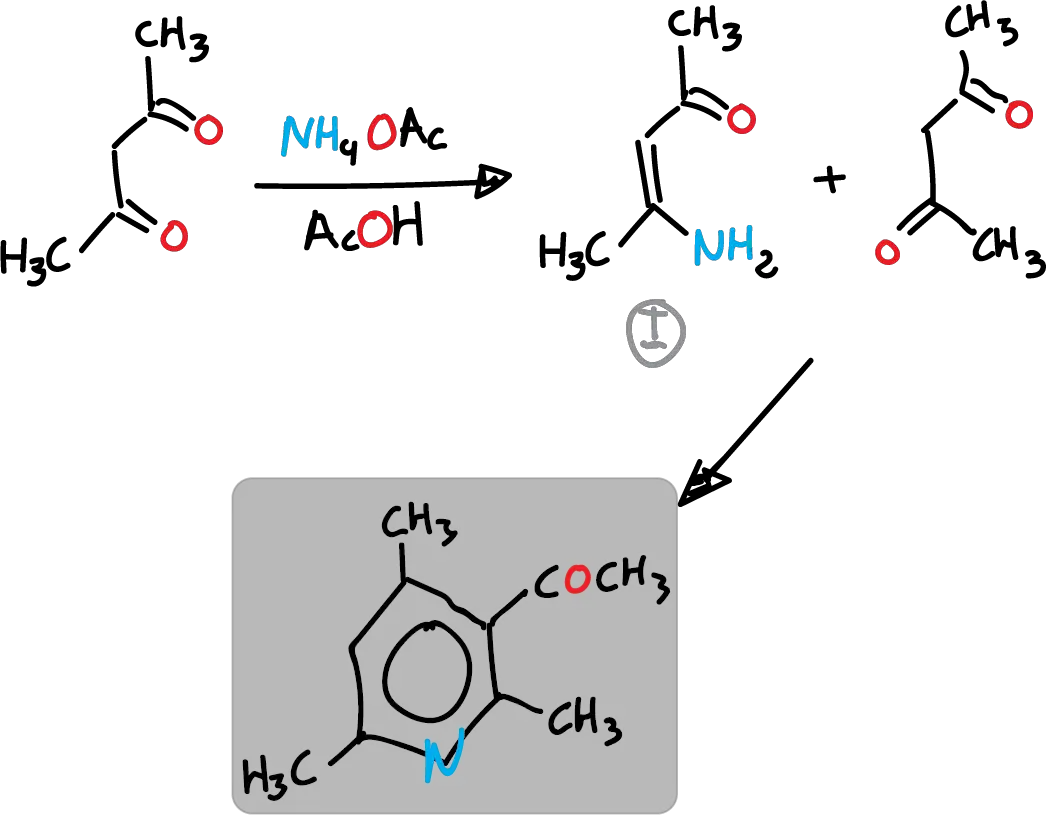
Route B, which is the cyclization of 1,5-diketones with ammonia, has rather limited application. However, a more versatile variant called Krönke synthesis is described below.
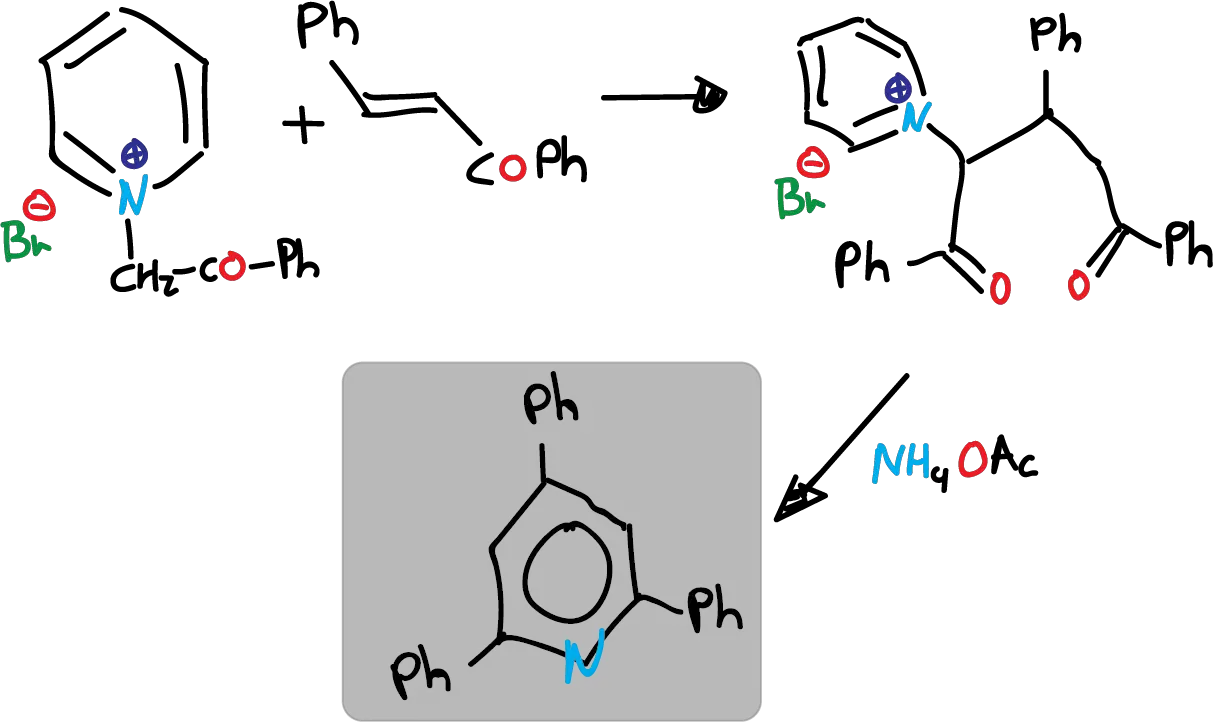
It involves the addition of a pyridinium ylide to an α,β-unsaturated carbonyl compound, leading to the formation of a 1,5-dicetone. This 1,5-diketone, is in the appropriate oxidation state to be directly cyclized.
Guareschi-Thorpe synthesis of 2-pyridones
It is used to prepare 2-pyridones, using cyanoacetamide as the nitrogenous component and with a 1,3-diketone or β-ketoester.
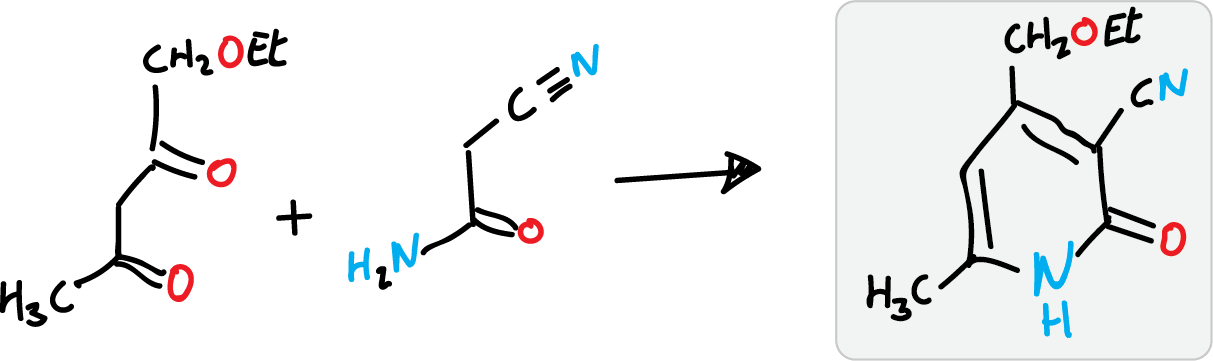
Other variants
Other synthesis methods have been described.
- For example, starting from ammonia and units of 5 carbon atoms (such as glutaconic aldehyde).
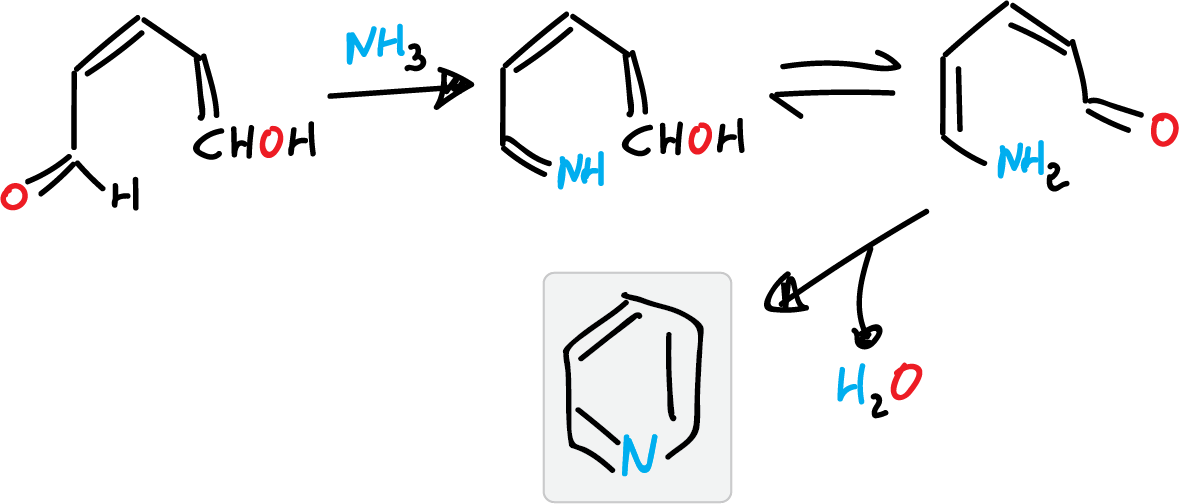
This reaction has no practical application since the glutaconic aldehyde is obtained by opening a pyridinium salt ring.
- Similarly, 1,5-dicarbonyl compounds react with hydroxylamine (NH2OH).
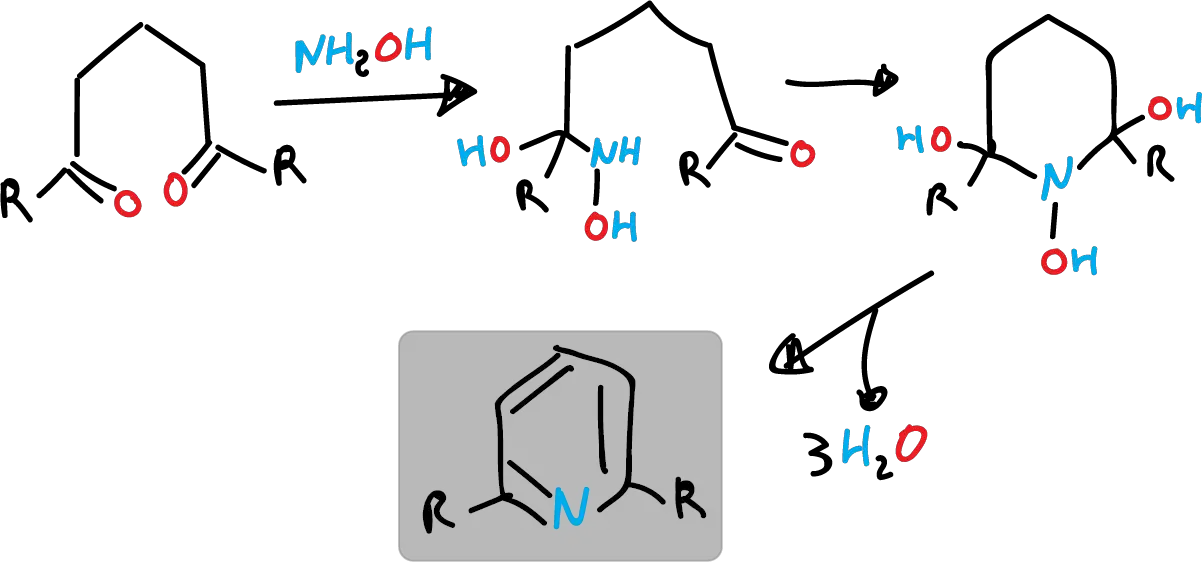
Synthesis of substituted pyridines
The synthesis of substituted pyridines, instead of starting from pyridine, is obtained from the various aliphatic compounds described above, suitably substituted. This is because electrophilic aromatic substitution (SEAr) is not favored on the pyridine ring. For electrophiles frequently attack basic nitrogen.
However, there are a wide variety of reactions that give pyridine and derivatives.
Chemical properties of pyridines
Pyridine is the heterocyclic compound most similar to benzene. It has a high resonance energy and its structure closely resembles that of benzene. Naturally, the presence of nitrogen gives it special characteristics. The electron pair in the plane of the ring provides a center susceptible to protonation and alkylation.
Many of the chemical properties of pyridine will therefore be that of a tertiary amine and the aromatic sextet is not involved in these reactions.
As mentioned, the characteristic reaction of benzene, electrophilic aromatic substitution (SEAr), is not evident in pyridines.
First of all, the product of the reaction of a pyridine with an electrophile is one in which the latter is coordinated with the electron pair of the nitrogen. Therefore, it hinders substitution on the carbon atoms of the ring, i.e., the initial formation of a pyridinium salt is favored.
For example, the action of dinitrogen pentoxide (N2O5) on pyridine, under neutral conditions gives:
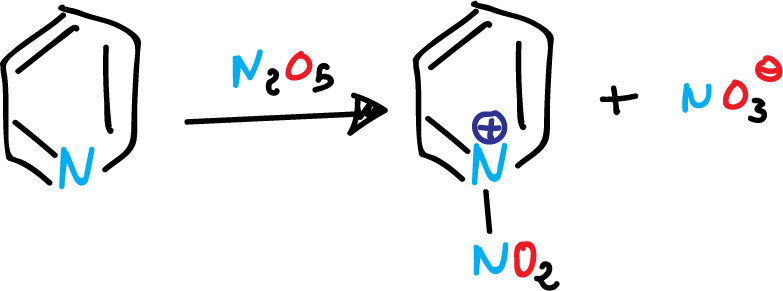
Nitration of the pyridine at position 1
However, when the C2 and C6 positions of the pyridine ring are substituted by bulky groups, coordination with the nitrogen is sterically prevented. Therefore, substitution on the ring can occur under relatively mild conditions.
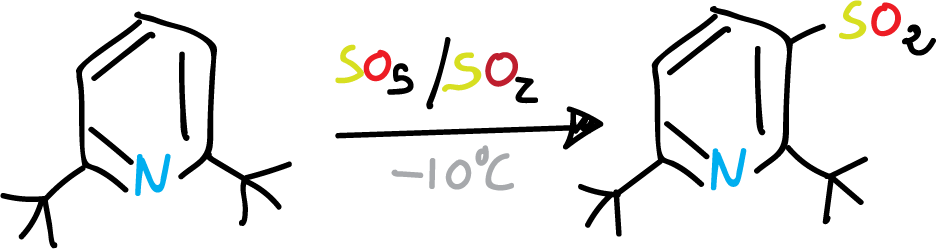
Sulphonation of pyridine with bulky groups at C2 and C6
SEAr in the pyridine and pyridinium ions preferentially occurs at the C3 position. This is due to the corresponding intermediates in the transition state, as indicated in the scheme.
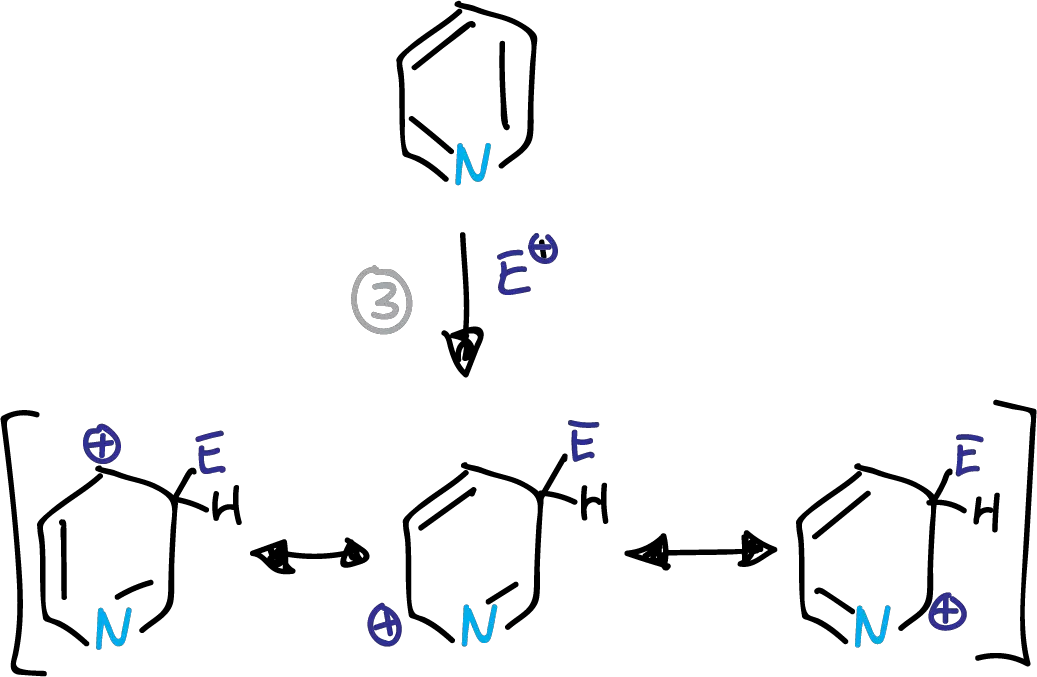
Intermediates in the electrophilic substitution of pyridine at the C3 position.
Whether the substitution occurs at C2 or C4, we find a disadvantaged form which is highlighted in the figures.
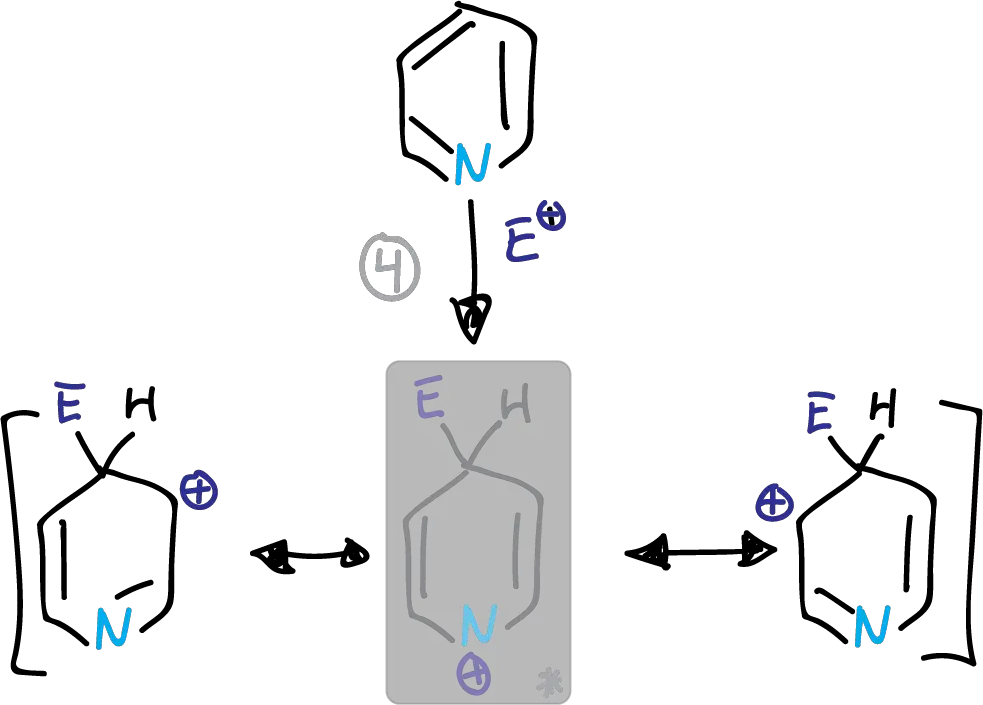
Intermediates in the electrophilic substitution of pyridine at the C4 position
This structure has a partial positive charge on the electronegative nitrogen. This causes the transition state energy to increase. The substitution is therefore preferably at the C3 position.
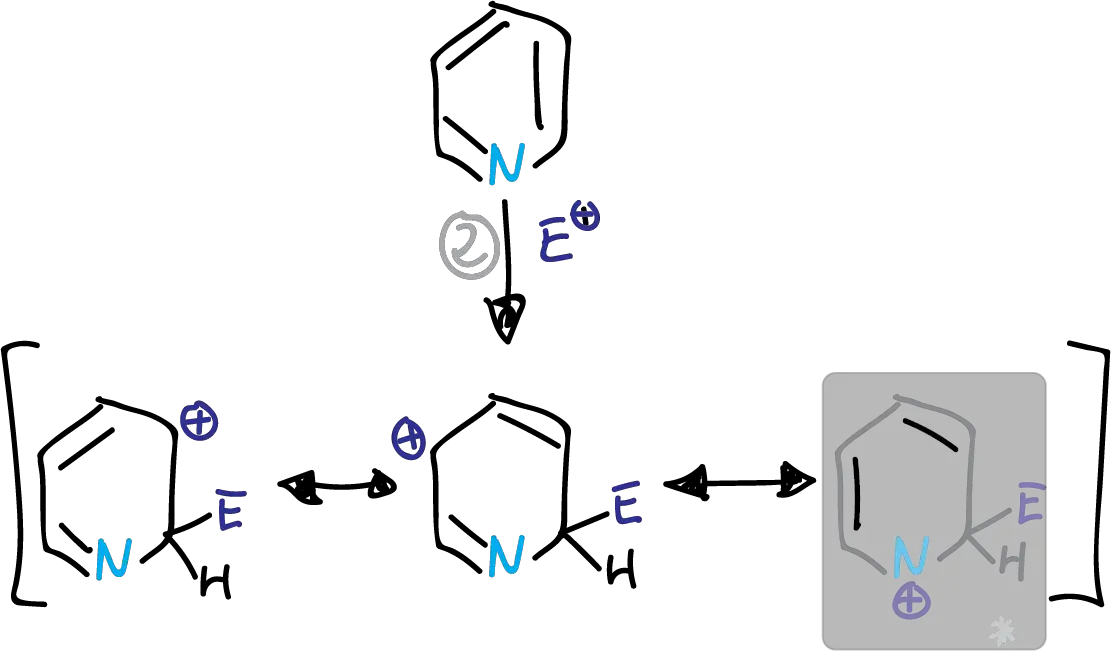
Intermediates in the electrophilic substitution of pyridine at the C2 position
It has been observed that alkyl substitutions activate pyridine, favoring elecrophilic substitution. For example in nitration.
To nitrate pyridine, vigorous conditions (H2SO4—SO3 /NO3K, 300 ºC) are required to give 3-nitro pyridine, with low yield.
A single methyl group is not sufficient to activate the pyridine, and the nitrant mixture rather oxidizes it. However, if we introduce several methyls, the reaction is easier.
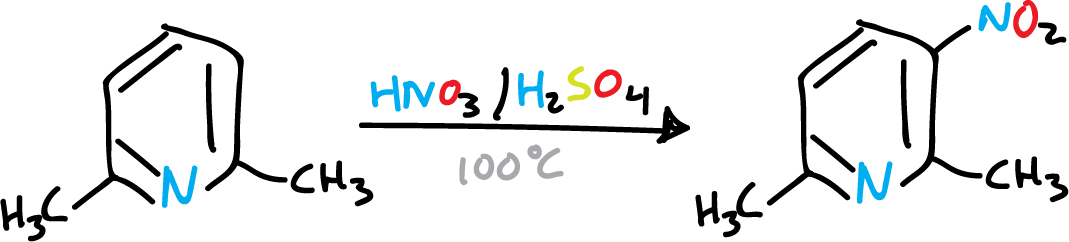
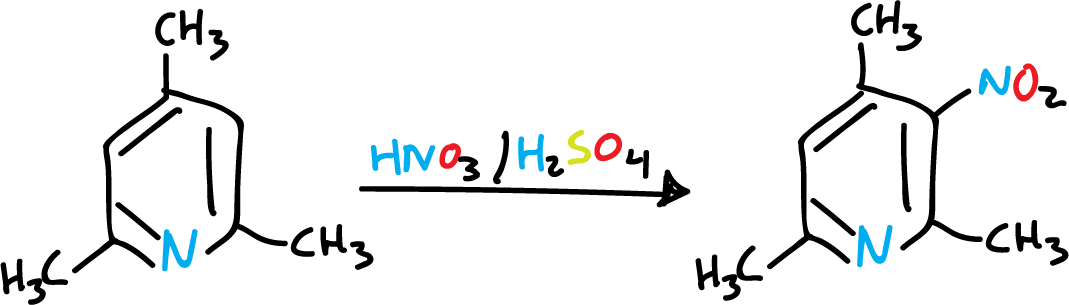
However, in a general way, the nitrogen atom of the heterocycle is still the predominant orienting influence in pyridines, as can be seen in the following sulfonation.
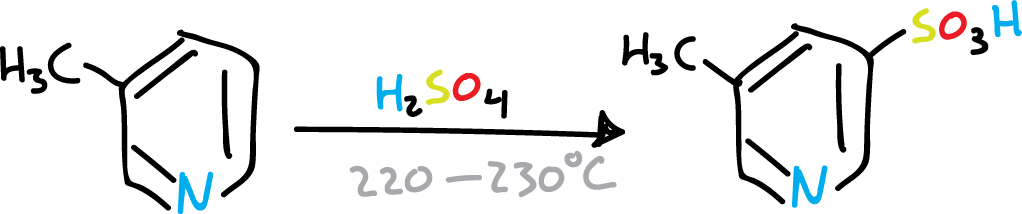
Sulphonation of pyridine.
Subsequent substitution of halogenated pyridines is also controlled by the heterocyclic center.
When the pyridine substituent is an amino group, these are the ones that exert the dominant orienting effect, facilitating substitution.
Thus, an amino group in C2 leads to C5 (para– director).
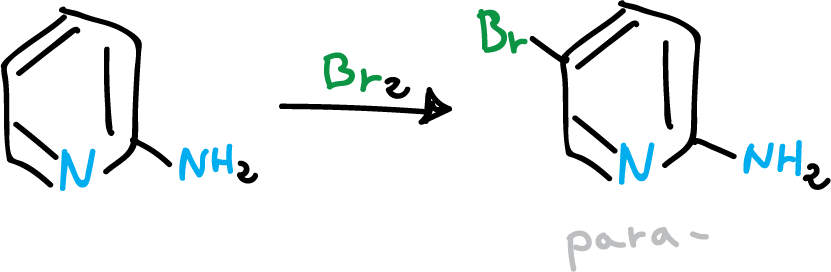
If the amino group is in C3 it directs C2 and if it is in C4 it directs C3 (ortho– director).

Bromination of the pyridine in the para position.
Also, C3 hydroxyl groups predominate in the orientation.
fig-22
If the hydroxyl is at C2 or C4, these structures exist predominantly in pyridone form, and as expected this influences their reactivity.
For example, 2-pyridones preferentially undergo attack in the C3 position, which contrasts markedly with the behavior of the 2-alkoxy derivatives, in which they are substituted at C5.
fig-23
The pyridine N-oxides can also undergo electrophilic attack at the C3 and C4 position depending on whether the reaction is caRied out with a free base or with the conjugate acid. Nitration is caRied out by attack on the free base.
fig-24
Thus, it is caRied out at position C4. When this position is occupied, nitration does not take place.
Sulfonation occurs with difficulty at the C3 position.
fig-25
When alkyl groups are present, they are activated, as in the case of pyridine, and here too the orienting effect of the heterocycle predominates.
In contrast, with amino substituents the order of directional power appears to be: NR2 > N⊕—O⊖ > NHCOR.
fig-26
The N-oxide groups have higher directing power compared to the alkoxide substituents in the oxidized products.
fig-27
However, the hydroxyl groups at C3 are quite strong (compared to N-oxide) and their influence predominates, as in the case of pyridine.
fig-27
2-Hydroxypyridine-N-oxide (N-hydroxy-2-pyridone) undergoes bromination to give the 3-brominated derivative and upon nitration, a substitution at the C5 position is observed.
fig-28
4-Pyridones and N-hydroxy-4-pyridones behave as expected and usually undergo substitution at C3 and C5
It is evident that pyridines and their N-oxides lack the necessary reactivity to interact with weaker electrophiles of the type generated in the Friedel-Crafts and Vilsmeier–Haack reactions (formylation with Ph(Me)NCHO and POCl3).