Introducción
A continuación, se describen las reacciones periciclicas, desde un punto de vista de sus aplicaciones. Es decir, veremos ejemplos prácticos de estas reacciones dirigidos a demostrar que las reacciones pericíclicas se dan y preferentemente frente a la alternativa no pericíclica. Además, se aborda desde un punto de vista experimental y no teórico. Tratamos de obtener pruebas que justifiquen la existencia de reacciones pericíclicas desde un punto de vista experimental.
Las reacciones pericíclicas presentan dificultad de estudiarlas experimentalmente puesto que no se pueden aislar los intermedios de reacción. Por tanto, lo que haremos experiemntalmente es comprobar que las alternativas no concertadas no se dan, por ejemplo demostrando que la reacción no se da por una vía radicalaria o que no se da por un intermedio iónico, etc.
Además, normalmente se estudia experimentalmente la estereoquímica de estas reacciones, debido a que deben de ser estereoespecíficas. Si se da una mezcla de productos de reacción cuando debería de darse un único estereoisómero, es una pista de que esa reacción no transcurre por un mecanismo pericíclico.
Reacciones de cicloadición
Comenzamos con las reacciones de cicloadición que dan lugar a distintos tamaños de ciclos.
Formación de anillos de tres miembros
Para obtener ciclos de tres miembros uno de los componentes tiene que actuar como un sólo átomo mediante una interacción de tipo ω. Las reacciones más simples que generan ciclos de 3 miembros son las reacciones con carbeno (en su estado singlete o triplete). Las reacciones en estado triplete no pueden ser reacciones pericíclicas puesto que es un radical y por tanto actuará mediante una reacción de tipo radicalario. Pero en carbenos singlete el par de electrones se podrá representar como un par de electrones sobre un orbital de tipo sp2, con dos substituyentes adicionales.
De manera que experimentalmente se observa que si la reacción se hace enpresencia de luz que pueda actuar como triplete, la reacción da mezcla de productos de reacción con la estereoquímica de los productos, lo cual demuestra que transcurre mediante un mecanismo radicalario. Pero si se lleva a cabo en ausencia de radicales se mantiene la esteroquímica del doble enlace, si este tenía un cierta estereoquímica se mantendrá en el ciclo de tres miembros. Si es un sistema de 4 electrones uno de ellos tiene que actuar antarafacialmente, lo que no es difícil en un carbeno, puesto que puede producirse una interacción de este tipo:
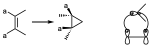
Que será una interacción antarafacial sobre el carbeno con una geometría muy sencilla, se conocen ejemplos en los que se conserva la estereoquímica del doble enlace y por tanto se piensa que son reacciones de tipo pericíclico en las que el carbeno actúa de forma antarafacialmente.
Reacciones quelotrópicas
Se conocen otros muchos ejemplos en los que se conserva la estereoquímica de la reacción, suelen ser reacciones de cicloreversión en lugar de cicloadición, por ejemplo por calentamiento de este tipo de estructuras se producen cicloreversiones:
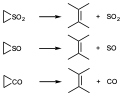
Se produce ese doble enlace y se conserva la estereoquímica de la reacción. Esto nos hace pensar que pueden ser reacciones de tipo pericíclico, pero como no se puede hablar de la estereoquímica sobre el grupo SO2, CO o SO que se forma, no se puede demostrar explícitamente que se trata de una reacción pericíclica.
Formación de anillos de cuatro miembros
Para formar anillos de cuatro miembros por reacciones de cicloadición, en lo primero que pensamos es en reacciones π2+π2, pero sabemos que esto está prohibido térmicamente, sin embargo fotoquímicamente si se puede producir, de manera que hay métodos muy sencillos de formar anillos de 4 miembros.

Sin embargo, se encontró unas reacciones térmicas que presentaban unas características interesantes que en un principio hizo pensar que se trataba de reacciones de tipo pericíclico y que después se ha demostrado que son radicalarias y que se detallan ya que nos muestran herramientas a emplear para demostrar si un mecanismo de reacción va por un mecanismo u otro.
En este tipo de reacciones se producía la cicloadición de sistemas diénicos pero por reacciones de cicloadición 2+2 es decir, actuaba uno de los dos dobles enlaces o también reacciones de sistemas diénicos con olefinas que estuvieran substituidas por halógenos.
Concretamente, se han estudiado los derivados clorados y fluorados 1,1-difúor,2-2 dicloro etitleno con butadienos. Se encontró que se obtenían de los posibles productos de reacción, exclusivamente unos frente a otros. Es decir, si partimos de una reacción de cicloadición 2+2 y se hacía reaccionar 1,2 butadieno:
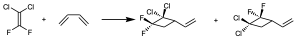
Se obtenían exclusivamente de las dos posibilidades de orientación de formación del sistema cíclico se obtenía sólo la segunda. Además, si el sistema diénico tenía una estereoquímica determinada (dienos substituidos), y presenta una estereoquímica cis o trans sobre los dobles enlaces, el producto de reacción conserva la estereoquímica del doble enlace.
Por ejemplo, si se hace reaccionar este sistema con estos dos compuestos, a partir de uno se forma un compuesto y a partir del otro, un segundo.
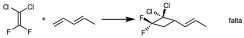
Estas características sobre la estereoquímica de la reacción y sobre la regioquímica que se da en la primera reacción, fue lo que hizo pensar que podía tratarse de reacciones de tipo pericíclico que violaran las reglas de selección, puesto que sería un caso de reacción 2+2 supra–supra.
Entonces, de una forma más exhaustiva se estudió la estereoquímica de estas reacciones, viendo todas las posibles reacciones que se dan frente a distintos dienos, de manera que se hizo reaccionar con estos compuestos:
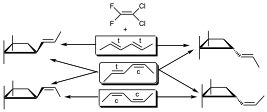
Se observó que a partir de ese mismo compuesto se obtenía una mezcla de los productos, y esta mezcla correspondía al isómero trans pero con el substituyente para arriba y el otro producto era aquel con el substituyente para abajo, con lo que se observa que puede existir la cicloadición de las dos formas.
Al hacer la reacción con el segundo se observó una mezcla de 4 productos, dos de ellos eran los obtenidos anteriormente y otros dos son aquel que tienen el substituyente en la forma cis.
Esto nos indica que la reacción no es estereoespecífica ya que a partir de cada uno de ellos se debería de obtener un único producto y se obtienen mezcla de dos y a partir del segundo deberían de obtenerse dos, uno con una configuración cis y otra trans. Sin embargo, se obtiene una mezcla de los cuatro.
Todo lo anterior, hizo pensar que es una reacción con unas características estereoquímicas peculiares, pero no es esteroespecífica y por lo tanto no puede ser una reacción pericíclica.
Se piensa que es una reacción radicalaria cuyo mecanismo para el caso del difluoro-dicloro es una reacción por pasos. Se produce primero el ataque de ese doble enlace a uno de los dos dobles enlaces del sistema diénico. Además, tiene lugar un ataque radicalario (se origina un diradical). De manera que en un primer paso obtendríamos un radical que por la presencia de ese doble enlace es un radical de tipo alílico que hace que la rotación alrededor de ese doble enlace no se pueda dar. Sin embargo, a lo largo del siguiente enlace puede haber rotación que evolucione al isómero que tiene el substituyente para abajo.
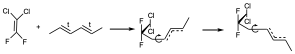
Por tanto, ahora puede ciclar en una posición o en otra. Si cicla en el primero dará lugar al producto primero de la reacción, en cambio si cicla el segundo dará lugar al segundo producto, pero ambos poseen la configuración trans en el doble enlace debido a la existencia de ese radical alilo, y de las dos posibilidades de ataque de regioquímica se obtiene sólo aquella en la que la posición de los átomos de flúor es la que se de en la reacción. Esto es debido a que un radical libre no es estable en las cercanías de dos átomos de flúor.
Sin embargo, si es estable en las cercanías de dos átomos de cloro, se obtiene preferentemente el ataque primero para dar lugar al radical unido al carbono que tiene los dos cloros que puede evolucionar y ciclar para dar lugar a los dos productos de la reacción.
Se han intentado diversos estudios experimentales para tratar de encontrar reacciones de cicloadición 2+2, aunque sea buscando la posibilidad de que uno de los componentes actuara antarafacialmente para ver si se puede dar en efecto una reacción 2+2.
Para que se produzca una interacción antarafacial en un componente con dos electrones, se tiene que producir una geometría de acercamiento como se indica:

Para que esto se produzca tenemos que tener los substituyentes sobre cada átomo de tal forma que los substituyentes sobre el sistema olefínico que tiene los orbitales π en el plano perpendicular sitúe esos substituyentes sobe el plano del papel. Y el sistema olefínico que se va acercando en posición vertical tiene que tener los substituyentes en el plano perpendicular al del papel, de manera que uno de los substituyentes va hacia dentro y otro hacia fuera.
Para que se pueda estudiar una reacción de este tipo experimentalmente tenemos que tener que los substituyentes sean adecuados para que se pueda observar una cierta estereoquímica.
Entonces, para que se pueda demostrar la existencia de una interacción antarafacial tiene que haber una cierta estereoquímica en los reactivos, que se tiene que conservar en uno de los componentes y tiene que producirse una inversión de la configuración en el que actúa antarafacialamente, y además como uno tiene que actuar de una manera perpendicular y acercando los grupos hacia el otro, esos grupos que se acercan deben de ser los más pequeños posibles para que se de la mínima interacción estérica posible.
Esto se ha intentado haciendo reaccionar estos dos compuestos.
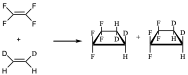
Esta es la posible reacción, en la que se puede observar la esteroquímica del componente que actúa antarafacialmente con los substituyentes más pequeños acercándose. De manera, que cuando se pare del isómero cis o trans, en ambos casos se obtiene la misma mezcla de productos en la que la configuración está repartida. Es decir, cuando hay 4 substituyentes en los dos sistemas olefínicos que intervienen, la reacción transcurre por mecanismo radicalarios puesto que se obtienen mezclas de productos. No se produce una reacción supra-supra puesto que está prohibida por simetría pero la supra–antara está prohibida por interacciones estéricas o por la tensión que supone el retorcimiento de uno de los componentes.
Al ver que no se podía observar la reacción antarafacial porque siempre hay alguna interacción aunque pongamos sustituyentes poco voluminosos. Se puede pensar en poner un reactivo que no tenga estos substituyentes, este sería el caso de las cetenas, donde no hay substituyentes sobre el oxígeno y si sobre el carbono y podemos intentar observar las reacciones de estas cetenas con olefinas adecuadamente substituidas para ver si al no tener uno de los átomos substituyentes, si se puede dar la reacción antarafacialmente.
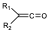
Se han obtenido características experimentales muy interesantes que hacen pensar que la reacción en realidad es una reacción pericíclica en la que se produce el ataque de uno de los componentes de una forma antarafacial.
Cuando se hacen reaccionar cetenas substituidas (con susbstituyentes distintos: uno voluminoso y otro pequeño) con una olefina también con substituyentes grandes y pequeños, se obtiene el ataque de la cetena de una forma determinada, que es la forma que se da lugar al sistema cíclico de 4 miembros en el que los substituyentes grandes quedan hacia un lado y los pequeños hacia otro.
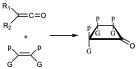
También cuando se hacen reaccionar cetenas convenientemente substituidas con ciclopentadieno en una reacción 2+2, se obtiene exclusivamente el producto endo con respecto al grupo grande.
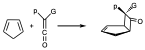
Estas peculiares características se podía deber a que reacciona la cetena de una forma antarafacial que explica la estereoquímica de la reacción. La olefina y la cetena la tendremos orientadas como se indica.

De las dos posibilidades de acercamiento de la cetena que puede ser con el grupo carbonilo hacia arriba o abajo, se dará aquella en la que la que se establezcan menos interaciones estéricas, si situamos el carbonilo hacia arriba, los substituyentes irán hacia abajo y como son más grandes que el carbonilo, se va a poder establecer una interacción estérica con los grupo grandes, por lo que el carbonilo por se la parte más pequeña de la cetena se situará con la orientación hacia los grupos grandes.
De los dos grupos que tiene la cetena se acercará hacia la molécula de etileno el pequeño, tal como la hemos dibujado. Esta debe ser la geometría de acercamiento hacia el ET para que se produzca una interacción antarafacial por parte de la cetena. Una vez que cicla evoluciona hacia un producto donde los grupos pequeños quedan todos del mismo lado.
Esto se ha visto con ejemplos de manera que si se pare de esta cetena y se hace reaccionar con cis-buteno se obtiene exclusivamente el producto que tiene el grupo más voluminoso hacia abajo y se mantiene la esteroquímica del cis-buteno. Con lo cual se puede decir que esa es una reacción pericíclica en la que uno de los componentes actúa de forma antarafacialmente.
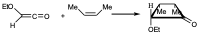
Formación de anillos de cinco miembros
Estamos viendo reacciones de cicloadición desde un punto de vista experimentalmente, se trata de ver ejemplos donde se pueda demostrar que existe una reacción pericíclica u otros casos donde se puede pensar que hay un cierto mecanismo pericíclico, de mostrar que no es así (van en contra de las reglas generalizadas de Woodward-Hoffmann).
Hay toda una serie de cicloadiciones que permiten formar anillos de 5 miembros por ejemplo las que se conocen con el nombre de cicloadiciones 1,3-dipolar que son reacciones π2+ π4 y por tanto deben de estar permitidas por simetría.
Existe una gran cantidad de reacciones de 1,3 dipolo (dialquenos) que reaccionan muy fácilmente para dar lugar a distintos tipos de sistemas heterocíclicos. Es una de las reacciones que más potencialidad sintética muestran par sintetizar heterocíclos.
Estas reacciones son estereoespecíficas y se tiene la idea de que son reaciones pericíclicas, lo que no quiere decir que el ET tenga que ser totalmente simétrico, que todos los enlaces que se tienen que formar estén igualmente formados que los que se tengan que romper estén rotos. Puede que haya un ET en el que una cierta carga esté más desplazada en una de las regiones que en otra y un de los enlaces esté más formado que otro. Pero esto no quita que la reacción sea concertada y se de en un sólo paso. Normalmente, se conocen muchos ejemplos de reacciones de cicloadición en la que participa algún 1,3-dipolo de los muchos que se pueden dar, como el óxido nitroso, los óxidos de nitrilo, ozono, óxido de carbonilo. Todos estos compuestos poseen estructuras de lo que se conocen como 1,3 dipolo y se caracterizan por poseer 4 electrones p en un sistema de 3 átomos, y pueden dar lugar a reacciones de cicloadición con olefinas y van a generar un anillo de 5 miembros. Así por ejemplo cuando se hace reaccionar una estructura de este tipo con un sistema de doble enlace:
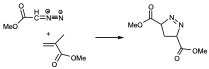
Estas reacciones se producen muy fácilmente a temperaturas bajas (en nieve carbónica) y dan lugar a los correspondientes heterociclos.
Otras reacciones de cicloadición, son por ejemplo las ozonolisis de alquenos. Los alquenos reaccionan con ozono también muy facilmente a baja temperatura (nieve carbónica o incluso menos) de tal manera que el mecanismo que se acepta es uno que presenta una serie de reacciones de cicloadición:
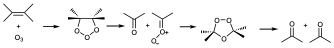
En una primera etapa se produciría una cicloadición de la olefina al ozono para dar lugar a lo que se conoce con el nombre de ozonido primario, que es una estructura muy inestable que produce una cicloreversión, pero en lugar de volver hacia atrás para dar de nuevo ozono, da lugar a un carbonilo y óxido de carbonilo que es también una estructura 1,3 dipolar. De manera, que si tenemos un óxido de carbonilo que es un 1,3-dipolo y un carbonilo que es un sistema p de dos electrones se puede producir otra cicloadición, cuando el sistema carbonílico tiene una estructura adecuada y no está muy impedido. En este caso la ciloadicón daría lugar a lo que se conoce con el nombre de ozónido, que ya son estructuras más estables y pueden incluso caracterizarse. Al tener el grupo peroxo, son estructura potencialmente peligrosas y lo que se suele hacer es destruirlas para dar lugar a dos productos que pueden ser iguales o distintos dependiendo de que la olefina fuera o no simétrica. Si la rotura se hace en un medio oxidante cada carbono pasa a su máximo estado de oxidación siempre que sea posible, es decir, a ácido carboxílico o a cetona si no se puede producir una oxidación posterior.
Hay una gran cantidad de pruebas que hacen pensar que es efectivamente ese mecanismo el que se produce, mecanismo Creege.
Se ha podido detectar el ozónido primario cuando la ozonolísis se hace a muy baja temperatura, por debajo de –100ºC y se estudia por RMN el producto de la reacción en el momento de producirse. Esto se hace realizando la reacción en un tubo dentro del aparato de RMN. En estas condiciones registrando el espectro antes de añadir el ozono y después registrando espectros cada cierto tiempo y aumentando la temperatura lentamente, se puede ver la evolución de los productos. Cuando se hace la reacción con este alqueno:
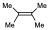
Se pueden reconocer fácilmente el pico correspondiente al reactivo, del pico correspondiente al producto primero de la reacción que es el ozónido primario y ese evoluciona posteriormente hasta la formación del ozónido secundario.
Hay otras pruebas que nos permiten por ejemplo detectar el óxido de carbonilo que es también una estructura muy inestable que no se puede aislar, pero si se pueden generar in situ por otros procedimientos que no sean la ozonolisis. O bien se pueden atrapar por algo que no sea el carbonilo que resulta de la reacción, para tratar de demostrar su existencia.
El oxido de carbonilo se puede generar en lugar de por ozonolisis, por reacciónes de fotólisis. Cuando se genera este óxido de carbonilo en presencia de aldehidos, por ejemplo en presencia de acetaldehido, se produce la reacción de manera que da lugar al ozónido correspondiente a la reacción de ese óxido de carbonilo con ese aldehido.
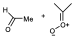
De tal manera que podemos decir que los óxidos de carbonilo si se forman reaccionan con los aldehidos para dar lugar a los oxónidos. Es una forma no necesaria de implicar que el óxido de carbonilo es un intermedio de lar reacción pero si se formara el óxido de carbonilo reacciona con los aldehidos.
Otra forma de detectar el óxido de carbonilo es partiendo de una estructura de una olefina en la que el óxido de carbonilo que se forme no pueda reaccionar con el carbonilo que también se forme en la reacción. Por ejemplo, en una olefina tetrasubstituida se formará un ozónido primario que evolucionará hacia la rotura para formar acetona y el óxido de carbonilo correspondiente.
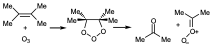
Si la reacción se hace en disolvente aprótico como puede ser cloruro de metileno ese óxido de carbonilo es incapaz de reaccionar con la cetona debido a impedimentos estéricos, en estas condiciones entonces lo que hace es reaccionar consigo mismo, dimeriza para dar lugar a este ozónido que es un producto paradójicamente absolutamente estable que se puede cristalizar, se le puede calcular el punto de fusión, etc.
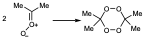
Es decir, que cuando hacemos una reacción de ozonolisis con una estructura en la que no se pueda producir la segunda reacción de cicloadición para dar lugar al ozónido, entonces el óxido de carbonilo si no puede reaccionar con nada más se dimeriza dando lugar a estructuras que son estables. La razón de porqué esas estructuras son tan estables teniendo como tienen dos grupo peroxo puede ser debido a la estabilidad del sistema cíclico de 6 miembros ya que lo que tenemos es una conformación en la que todos lo ángulos son tetraédricos y ángulos dihedros tienen una estructura alternada, etc.
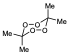
La reacción de 2,3 dimetilbuteno con ozono, en lugar de hacerla en diclorometano se hace en etanol, el óxido de carbonilo tampoco puede reaccionar con la cetona que se forma, pero ahora puede hacerlo con el disolvente, de manera que se daría lugar a esta estructura.
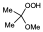
Este hidroperóxido es algo inestable pero se puede caracterizar de manera que demuestra en cierta forma que el óxido de carbonilo se forma. Ya que tanto este hidroperóxido con el dimero anterior sólo pueden proceder de la reacción del óxido de carbonilo.
Si se hace esa misma reacción en presencia de un aldehido, se obtiene el ozónido resultante de la reacción del óxido de carbonilo que se forma con el aldehido que hemos puesto.
Todo esto son pruebas más que concluyentes para pensar que efectivamente ese es el mecanismo que se da en una ozonolisis normal en la que las olefinas o no están muy impedidas o no son muy ricas en electrones. Que como vemos son una serie de dos cicloadiciones y una cicloreversión.
En general hay muchas pruebas experimentales que hacen pensar que las reacciones 1,3 dipolares son reacciones pericíclicas. Pero como siempre se proponen mecanismos alternativos. Teóricamente no puede ser un mecanismo radicalario pero sí un mecanismo dipolar, ya que los 1,3-dipolos son estructuras que tienen una separación de cargas importantes y pueden producir un mecanismo en el que se de un ataque en dos pasos. Un primer ataque de la zona de exceso de carga positiva de la olefina dando lugar a otro dipolo que puede cerrar posteriormente.
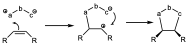
Si eso cicla por interacción electrostática de una forma muy rápida antes de que se pueda producir el giro alrededor de ese enlace, dará lugar al producto de ciloadición en el que se mantiene la estereoquímica de la olefína originaria.
Si por el contrario esa reacción no es tan rápida y da tiempo a que se pueda producir giro alrededor de ese enlace, entonces tendríamos una mezcla de los dos estereoisómeros.
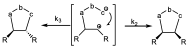
Firestone proponian este tipo de mecanismo y decía que no es que se produjera una reacción pericíclica sino que esa k2 es mucho más rápida que la k3 que da lugar al giro.
Es posible que dependiendo de ciertas variable se de un mecanismo u otro. Pero de forma general puede decirse que las reacciones de cicloadición 1,3-dipolar suelen ser reacciones pericíclicas, en algunos casos especiales donde existan efectos estéricos muy importantes puede darse el caso de que sean reacciones en dos pasos donde se produzca un ataque dipolar donde se produce un intermedio en el que se ha formado un enlace y el otro todavía está sin formarse.
Catedrático de Química Orgánica en la Universidad de Granada, con una larga trayectoria en Química Computacional, en modelado y diseño molecular.