Índice
Generalidades
Muchas moléculas orgánicas presentan la particularidad de tener la misma fórmula molecular, pero distintas propiedades fisicoquímicas, recibiendo el nombre de isómeros. En el siguiente esquema se resumen los tipos de isomería propios de moléculas orgánicas:
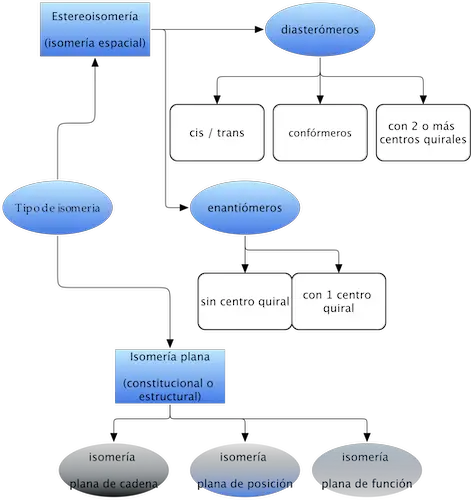
Isomería plana (constitucional o estructural)
Los isómeros planos se diferencian en la conectividad (forma en que se unen entre sí los átomos), poniéndose de manifiesto estas diferencias cuando los representamos en el plano. Se clasifican en tres tipos:
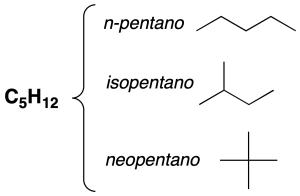
Isomería de cadena
Presentan distinta cadena principal.
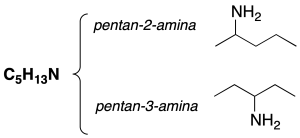
Isomería de posición
La posición del grupo funcional en la cadena es diferente.
Isomería de función
Estos isómeros presentan grupos funcionales distintos.

Estereoisomería (isomería espacial)
Dichos isómeros presentan la misma conectividad (igual número y tipo de enlaces), pero difieren en su configuración (la forma en que sus átomos están orientados en el espacio). La agrupación responsable de la estereoisomería se denomina unidad estereogénica.
Los estereoisómeros pueden ser de dos tipos:
Enantiómeros
Diasterómeros
Dos estereoisómeros que no son imágenes especulares. Se subdividen en:
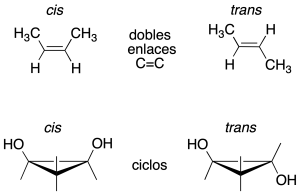
Isomería cis-trans
Cuando dichos isómeros no se pueden interconvertir mediante rotación en enlaces. Dos agrupaciones de átomos unidos a cada uno de los carbonos con un doble enlace, o anillo, que difieren en las posiciones con respecto al plano de referencia del doble enlace o del anillo.
Isomería conformacional
Cuando dichos isómeros se pueden interconvertir mediante una rotación en torno a enlaces sencillos, con más o menos restricciones.
Isómeros con más de 2 centros quirales
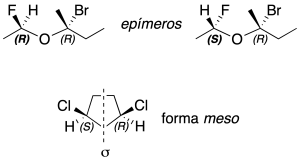
Una molécula con n carbonos quirales puede presentar un número máximo de 2n estereoisómeros, cuando no contiene planos de simetría (Le Bel y Van’t Hoff).
Si dos diasterómeros con 2 o más centros quirales se diferencian sólo en la configuración absoluta (véase convenio de Cahn-Ingold-Prelog) de un carbono quiral se dice que son epímeros; y si una molécula tiene centros quirales pero en su conjunto es aquiral, debido a la presencia de un plano de simetría, recibe el nombre de forma meso.
Convenio Cahn-Ingold-Prelog
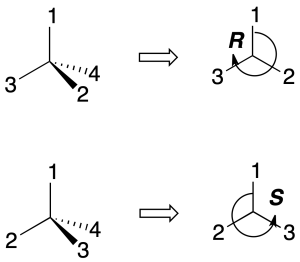
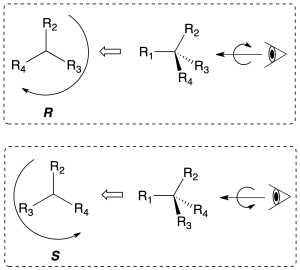
Para establecer la disposición de los grupos unidos a un carbono quiral de forma inequívoca se define el concepto de configuración absoluta. Para ello, se clasifican los sustituyentes del centro quiral según un orden de prioridad. Se gira el carbono de forma que el grupo de menor prioridad (4) quede hacia atrás del observador.
Se le asigna al carbono la configuración R(rectus), si dichos sustituyentes se disponen en el sentido horario, de mayor a menor prioridad (1-2-3). Contrariamente, se le asigna la configuración S (sinister) si en el sentido antihorario se disponen ordenados de mayor a menor prioridad (1-2-3), como se indica en la figura:
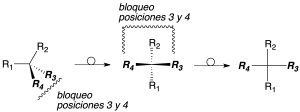
Para establecer la prioridad de los sustituyentes de un carbono quiral, se siguen las siguientes reglas secuenciales:
Regla 1
Los átomos unidos a un carbono quiral se clasifican según su número atómico.
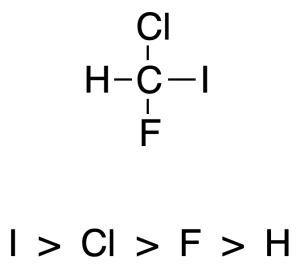
Regla 2
En el caso de que dos átomos unidos directamente al carbono quiral presenten el mismo número atómico, se consideran los átomos separados por dos, tres enlaces, etc., respecto del carbono quiral, hasta encontrar una diferencia.
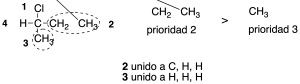
Regla 3
Si a lo largo de una cadena encontramos una ramificación sin haber establecido la prioridad, se compararán las cadenas de los grupos con mayor prioridad de la bifurcación y se establece la diferencia a partir de los átomos unidos a estos grupos.
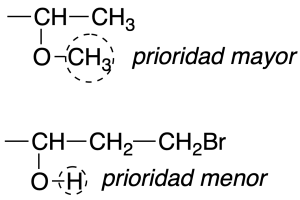
Regla 4
Cuando existen enlaces múltiples, se considera como si los enlaces fuesen saturados, de la siguiente manera:
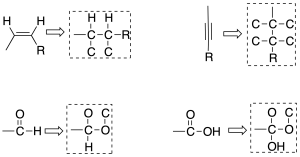
Regla 5
Si dos átomos son isótopos entre sí, presenta mayor prioridad el de mayor masa atómica. Por ejemplo 14C > 13C.
Regla 6
Si la diferencia entre dos sustituyentes radica únicamente en su configuración absoluta, el sustituyente de configuración R, tiene mayor prioridad que el S. Los carbonos quirales de este tipo reciben el nombre de carbonos pseudo-asimétricos y se anotan con las letras r y s minúsculas.
| Nº Prioridad | Función |
| 1 | -I |
| 2 | -Br |
| 3 | -Cl |
| 4 | -SO2R |
| 5 | -SOR |
| 6 | -SH |
| 7 | -F |
| 8 | -OPh |
| 9 | -OCH2Ph |
| 10 | -OMe |
| 11 | -OH |
| 12 | -NO2 |
| 13 | -NO |
| 14 | -NMe2 |
| 15 | -NHEt |
| 16 | -NHMe |
| 17 | -NH3+ |
| 18 | -NH2 |
| 19 | -COOH |
| 20 | -COPh |
| 21 | -COCH3 |
| 22 | -CHO |
| 23 | -Ph-Me |
| 24 | -C≡CR |
| 25 | -Ph |
| 26 | -C≡CH |
| 27 | -CMe3 |
| 28 | -CH=CHMe |
| 29 | -C6H11 |
| 30 | -CH=CH2 |
| 31 | -CH2Ph |
| 32 | -CH2-C≡CH |
| 33 | -CH2-CH=CH2 |
| 34 | -CH2-CHMe2 |
| 35 | -CH2-CH2-CHMe2 |
| 36 | -CH2-CH2-CH2-CH2-CH3 |
| 37 | –n-But |
| 38 | –n-Prop |
| 39 | -Et |
| 40 | -Me |
| 41 | -H |
Criterios para la asignación de la configuración de dobles enlaces
Los dobles enlaces C=C pueden presentar distintas orientaciones de los sustituyentes. Cuando sólo hay dos sustituyentes (en C1 y C2) diferentes al hidrógeno, los dos posibles isómeros pueden distinguirse mediante la notación cis / trans para indicar si ambos grupos se encuentran, respectivamente, a un mismo lado del doble enlace o a lados diferentes.
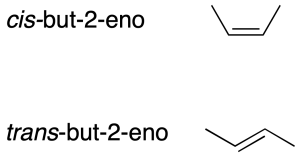
En alquenos más complejos esta forma de diferenciar los dobles enlaces no es válida.

El sistema de nomenclatura adoptado se basa en las reglas secuenciales de prioridad de Cahn-Ingold-Prelog visto anteriormente para los grupos unidos a cada carbono.
Si los sustituyentes de mayor prioridad se encuentran al mismo lado del doble enlace, se dice que éste tiene una configuración Z y si se encuentran al lado contrario del doble enlace se le asigna al doble enlace, la configuración E. La letra viene del término alemán Zusammen y Entgegen.
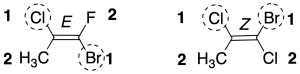
Este sistema de nomenclatura es más general que el cis / trans, cumpliéndose que los dobles enlaces de configuración cis son Z y los de configuración trans son E.
Magnitudes relacionadas con la isomería óptica
Cuando tenemos un producto quiral o una mezcla de distintos isómeros quirales hay una serie de magnitudes que sirven para indicar la pureza de una muestra:
Rotación específica [α]D25
La rotación específica se define como el ángulo de rotación óptica (a) observado, cuando un haz de luz de λ = 589 nm (línea D del sodio) pasa a través de una muestra en una celdilla de paso 0,1 m, concentración 1 g/dl (o bien 1 g/100 ml y celdilla 10 cm o 1 dm), y 25 ºC de temperatura.
![]()
![]()
Para dos enantiómeros esa rotación específica tiene el mismo valor absoluto, pero distinto signo.
Pureza óptica (p.o.)
La pureza óptica de una mezcla se define como el cociente entre la rotación observada de dicha mezcla dividido por la rotación óptica del enantiómero puro.
![]()
![]()
Exceso enantiomérico (e.e.)
El exceso enantiomérico expresa también la proporción relativa de enantiómeros (+) y (-) en una mezcla. El exceso del enantiómero predominante se calcula como porcentaje de la mezcla.
![]()
![]()
Relación entre estructuras orgánicas
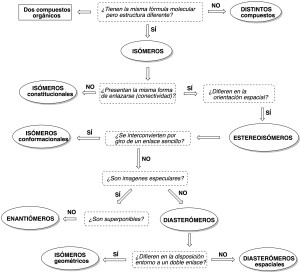
Cuando se pregunta cual es la relación entre dos estructuras orgánicas debemos responder a las siguientes cuestiones sucesivamente que se muestran en el siguiente diagrama de decisión:
En la siguiente figura se resumen las diferentes formas de representación de moléculas: proyección de caballete; representación de Cram (trazos en cuña); representación de los ciclos en el plano como polígonos regulares; proyecciones de Fischer, Haworth y Newman.
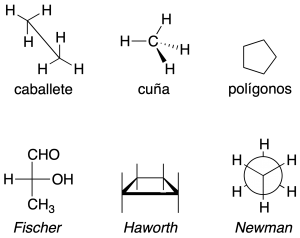
Una misma molécula la podemos representar de cualquiera de esas formas y no siempre la analogía es evidente; es por ello que necesitamos poseer la visión necesaria para la interconversión entre ellas. La manera más fácil de encontrar la relación entre distintos estereoisómeros es determinar las configuraciones absolutas de todos sus carbonos quirales y después, comparar los resultados obtenidos. Para poder comparar estructuras en diferentes representaciones, quizás lo mejor sería interconvertirlas todas a la misma representación.
Aun así, no es tan simple como parece; por ejemplo, la proyección de Fischer de uno de los enantiómeros de una molécula con un solo carbono quiral (el ácido (R)-2-hidroxipropanoíco o ácido (R)-láctico) se puede representar de 8 maneras distintas:
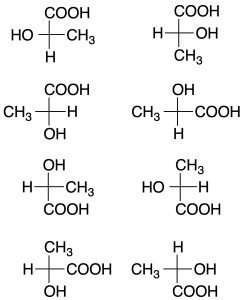
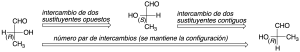
En dicha proyección, si se intercambian de posición dos sustituyentes, cambia la configuración del carbono quiral. Si se realiza un número par de permutaciones con dos sustituyentes, se mantiene la configuración del carbono quiral. Sin embargo, si el número de permutaciones entre dos grupos es impar, cambia la configuración del carbono quiral.
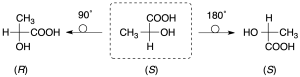
También hay que tener en cuenta que en dicha representación se puede girar la molécula 180º sobre el plano que la contiene, pero no 90º pues, en este caso, cambiaría la configuración:
Isomería conformacional
Llamamos confórmeros a las distintas disposiciones espaciales que pueden adoptar los átomos de una molécula como resultado del giro de uno o varios enlaces simples en la misma.
Se suelen visualizar más fácilmente mediante la proyección de Newman.
Proyección de Newman:
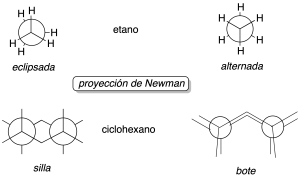
Ejemplo de cómo obtener una conformación a partir de otra
Por ejemplo, las conformaciones del 1,2-difluoroetano se obtendrán a partir de una conformación determinada y manteniendo fijo uno de los carbonos, ir girando el otro hasta obtener una disposición coincidente con la primera. Si partimos de la conformación eclipsada a, y mantenemos fijo el carbono más próximo al observador, se va girando el más alejado en pasos de 60º.
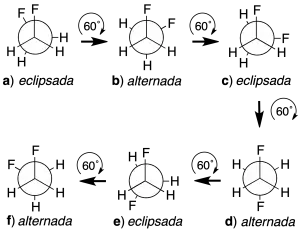
Como resultado, se obtienen las conformaciones a–f. Para nombrar las conformaciones se siguen los siguientes criterios:
- Cuando los grupos más voluminosos forman ángulos diedros inferiores a 90º, como ocurre con los dos átomos de flúor en la conformación eclipsada a, se utiliza el prefijo sin.
- El prefijo anti se utiliza cuando los grupos más voluminosos forman ángulos diedros superiores a 90º, como ocurre en la conformación alternada d.
- Si los grupos más voluminosos se sitúan en planos diferentes, como ocurre en b o en f, se usa la terminación clinal
- Utilizamos la terminación periplanar cuando los dos grupos más voluminosos se sitúan en un mismo plano (ángulo diedro 0º) como ocurre en a o d.
Así, las conformaciones a, c y e son eclipsadas, mientras que las conformaciones b, d y f son alternadas. La e es equivalente a la c y la conformación f a la b. En el caso de disposiciones sinclinales se utiliza como sinónimo el término gauche (del francés torcido). Según estas normas, se pueden nombrar las conformaciones de la siguiente manera:
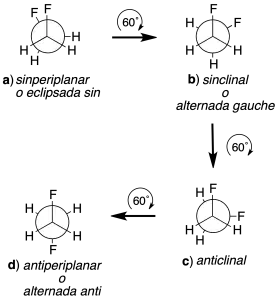
Ejemplo de como obtener el confórmero predominante
Otra cuestión adicional del análisis conformacional es decidir cuál es el confórmero predominante de una sustancia determinada. Por tanto, puesto que todos los confórmeros son interconvertibles entre sí por giro de un enlace simple, todos los confórmeros estarán presentes en el equilibrio. Como consecuencia, su abundancia relativa dependerá de las estabilidades relativas de los mismos. Así, para determinar cuál es el más estable, será aquel que presente menos interacciones estéricas y polares.
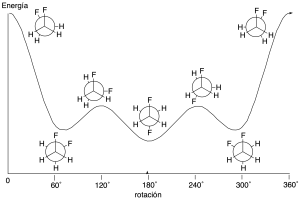
De forma similar se analizarán los compuestos cíclicos (especialmente ciclohexanos) aunque, en este caso, las interacciones más importantes son las 1,3-diaxiales (al no existir conformaciones eclipsadas). Éstas suelen estar cuantificadas en términos energéticos y permiten, incluso, calcular la distribución de confórmeros en el equilibrio.
Catedrático de Química Orgánica en la Universidad de Granada, con una larga trayectoria en Química Computacional, en modelado y diseño molecular.