Written by José | Last Updated on 2 meses
Generalidades
La síntesis orgánica tiene por objeto la construcción de moléculas de mayor complejidad a partir de estructuras más sencillas. Este proceso supone el encadenamiento de reacciones a partir de un sustrato determinado, para llegar a la obtención de la molécula objeto de la síntesis.
Índice

En moléculas con varios grupos funcionales, los procedimientos sintéticos se convierten, prácticamente en un “traje a medida”para cada caso, ya que son tantos los tipos de reacciones y las formas de obtener una molécula (grupo funcional), que la adaptación de las reacciones de la Química Orgánica a compuestos multifuncionales resulta ser un arduo trabajo, en el que la creatividad y la intuición tienen un papel destacado. Por eso, no es de extrañar que algunos autores hablen de la síntesis orgánica en términos de arte.
En cursos básicos de Química Orgánica, la síntesis orgánica se sustenta en el uso de las reacciones que se van aprendiendo con el desarrollo de los programas, para la preparación, en pocas etapas, de moléculas con un número limitado de grupos funcionales y con esqueletos carbonados no demasiado grandes.
A pesar de estos objetivos relativamente modestos, los estudiantes encuentran serias dificultades para abordar estos ejercicios, ya que es necesario relacionar múltiples conceptos y adquirir una visión global de los conocimientos adquiridos.
Tipos de síntesis: cuando se requiere realizar una síntesis multietapa, se puede llevar a cabo de dos formas:
Síntesis lineal: se sigue una secuencia de reacciones, de forma que el producto de una etapa sirve como reactivo de la etapa siguiente.
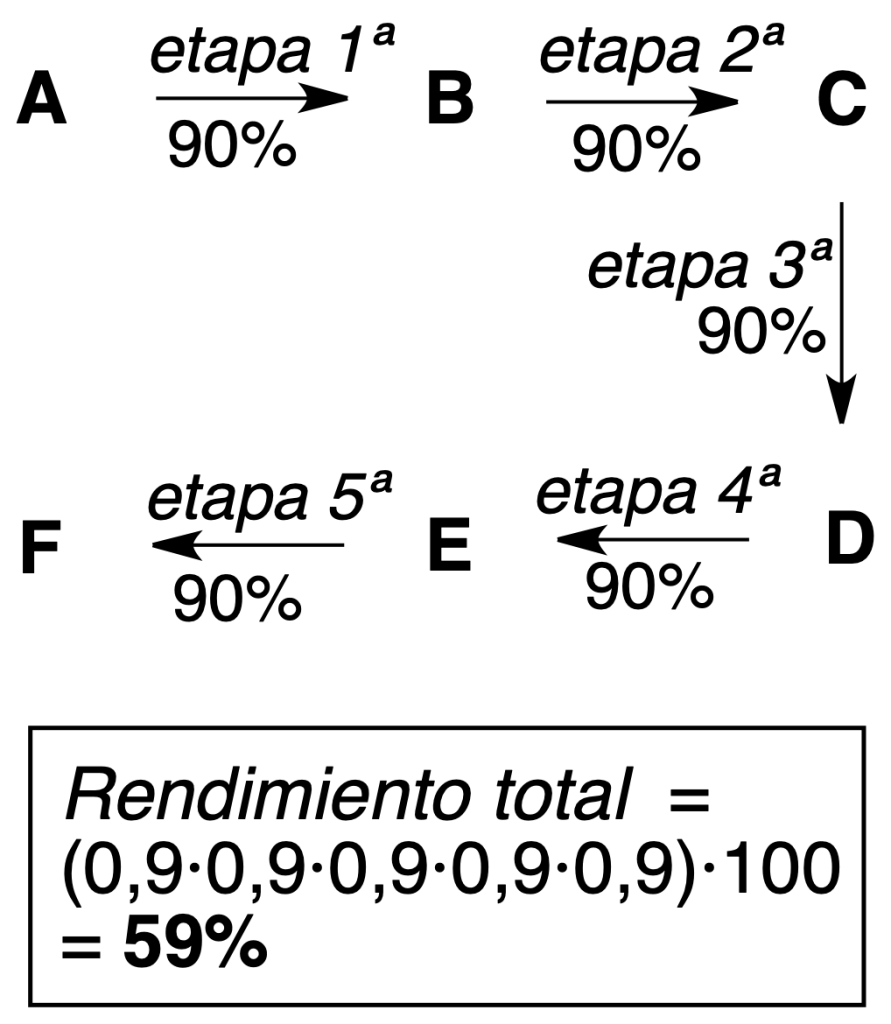
Síntesis convergente: el producto final (E) se obtiene por reacción entre dos moléculas (C y M) que provienen de dos rutas sintéticas diferentes.
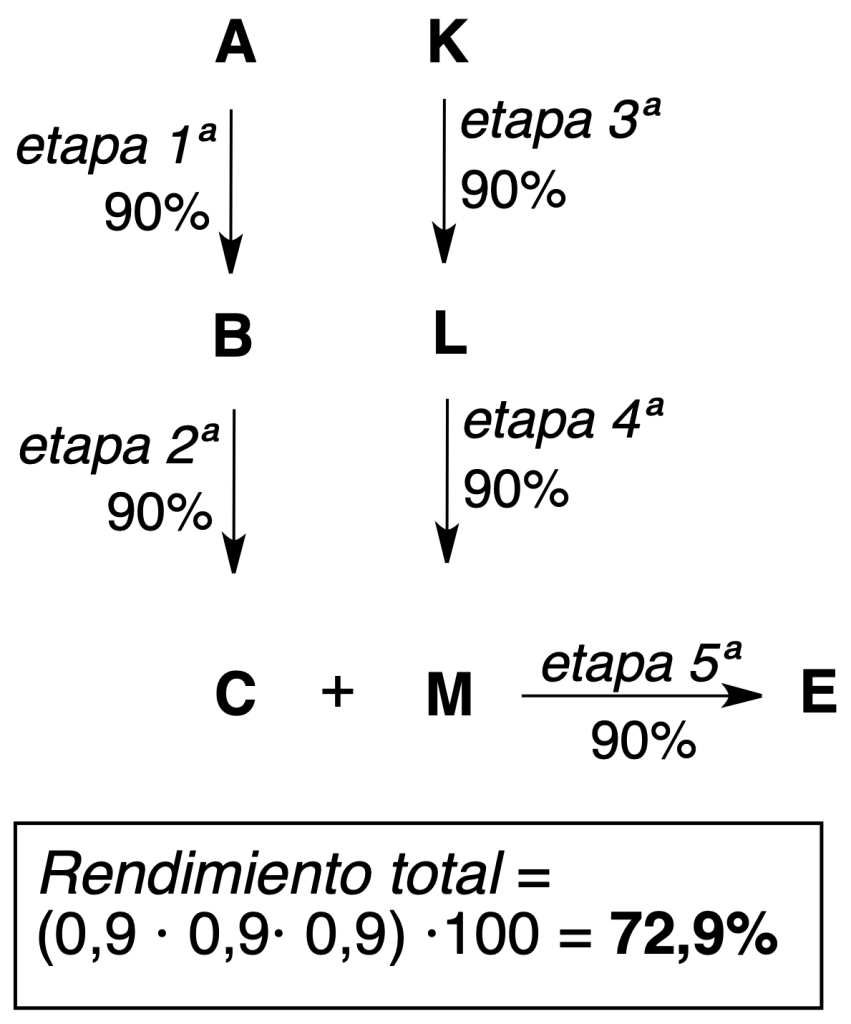
Como se ve en los ejemplos anteriores de procesos consistentes en 5 etapas, con la síntesis convergente se obtienen mayores rendimientos teóricos (más eficiente).
Conceptos útiles para clasificar reactivos y reacciones:
A continuación se definen ciertos conceptos que permiten clasificar reactivos o reacciones de la siguiente manera:
Quimioselectividad: se aplica a un reactivo que actúa preferentemente sobre un grupo funcional determinado de una molecula, con respecto a otro u otros grupos funcionales presentes en dicha molécula.
Regioselectividad: en una reacción, la formación o rotura de un enlace se produce en una dirección determinada de todas las posibles.
Estereoselectividad: formación preferencial de un estereoisómero sobre otro, en una transformación química.
Estereoespecificidad: una reacción se considera estereoespecífica cuando, partiendo de sustancias que difieren en su configuración, se obtienen productos que son entre sí estereoisómeros.
Desde otro punto de vista, los tipos de reacciones que se utilizan cuando se plantea una síntesis orgánica determinada, se pueden agrupar en dos grandes categorías que abordamos en los dos apartados siguientes.
Interconversión de grupos funcionales (IGF)
Dentro de la síntesis orgánica, un apartado importante lo constituye la IGF. En moléculas con una sola función esta tarea no supone una gran dificultad, pues basta con tener unas nociones básicas de la química de los grupos funcionales.
Cuando una molécula tiene más de un grupo funcional, existen dos opciones:
Uso de reactivos selectivos para unos determinados grupos funcionales con respecto a otros (quimioselectividad).
Uso de grupos protectores que impidan que reacciones posteriores alteren un determinado grupo funcional.
Grupos protectores
Ejemplos
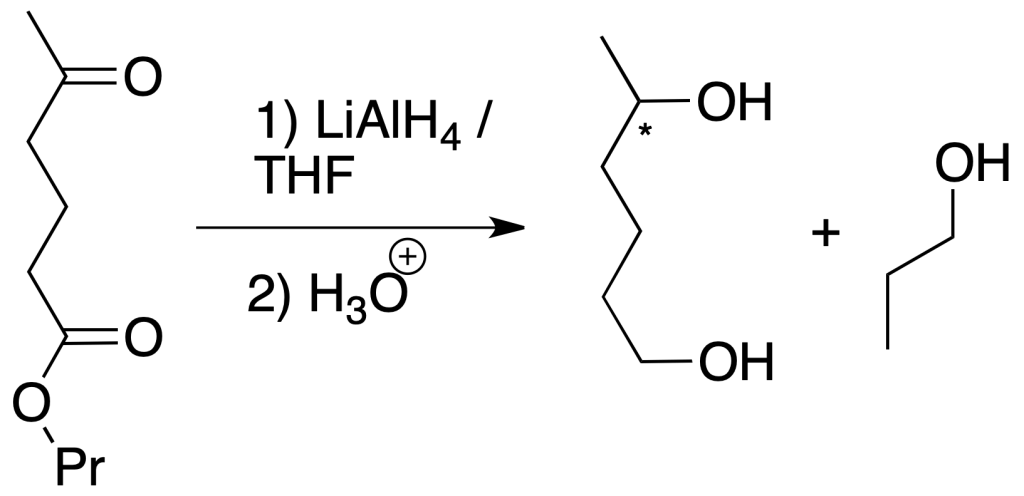
Si la siguiente molécula se trata con un agente reductor, tanto la función éster como la función cetona son susceptibles de ser reducidas.
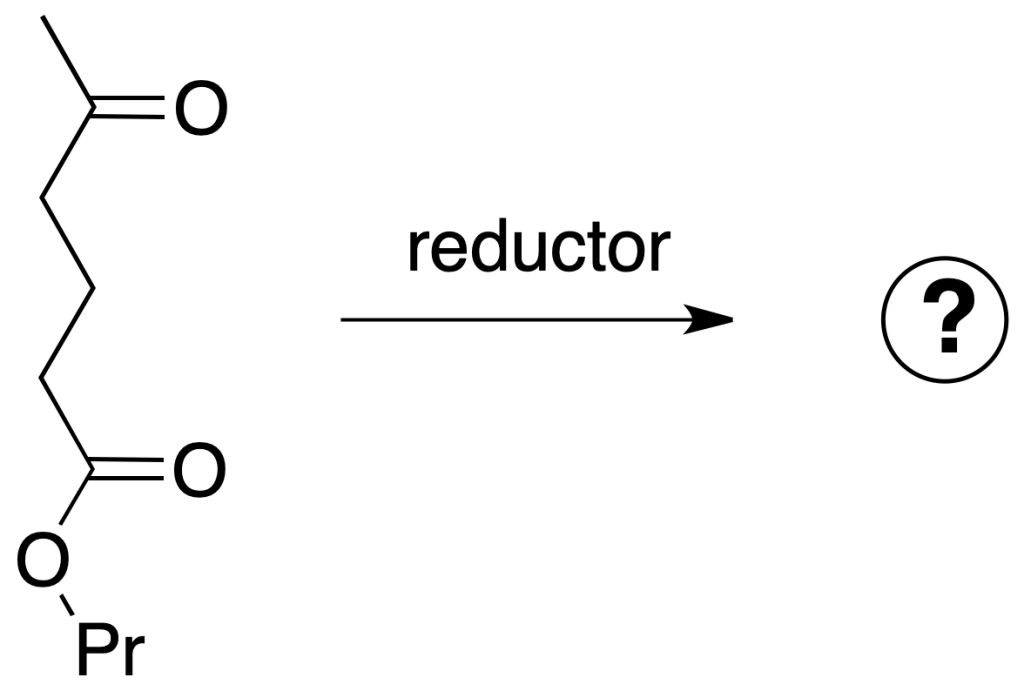
Se pueden plantear tres casos:
Reducción de ambos grupos (cetona y éster): para la primera opción, se puede emplear el LiAlH4. Este reactivo es un reductor fuerte que reduce compuestos carbonílicos hasta alcoholes y transforma ésteres en alcoholes primarios. En este caso, no se discrimina entre ambas funciones, que se reducen de forma simultánea (la reacción no es quimioselectiva).
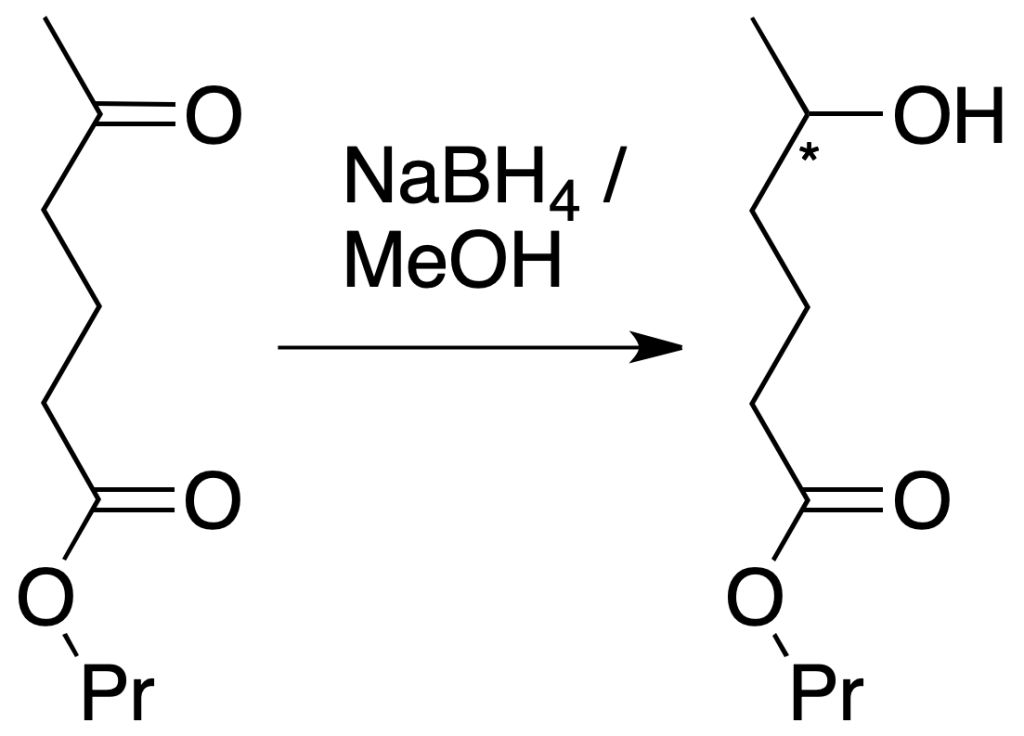
Reducción sólo de la cetona: si por el contrario, se usa NaBH4, se reduce la función cetona, mientras que permanece inalterado el grupo éster. En este caso, el NaBH4 reduce selectivamente al grupo carbonilo de la cetona, en presencia del éster que no reacciona (la reacción es quimioselectiva).
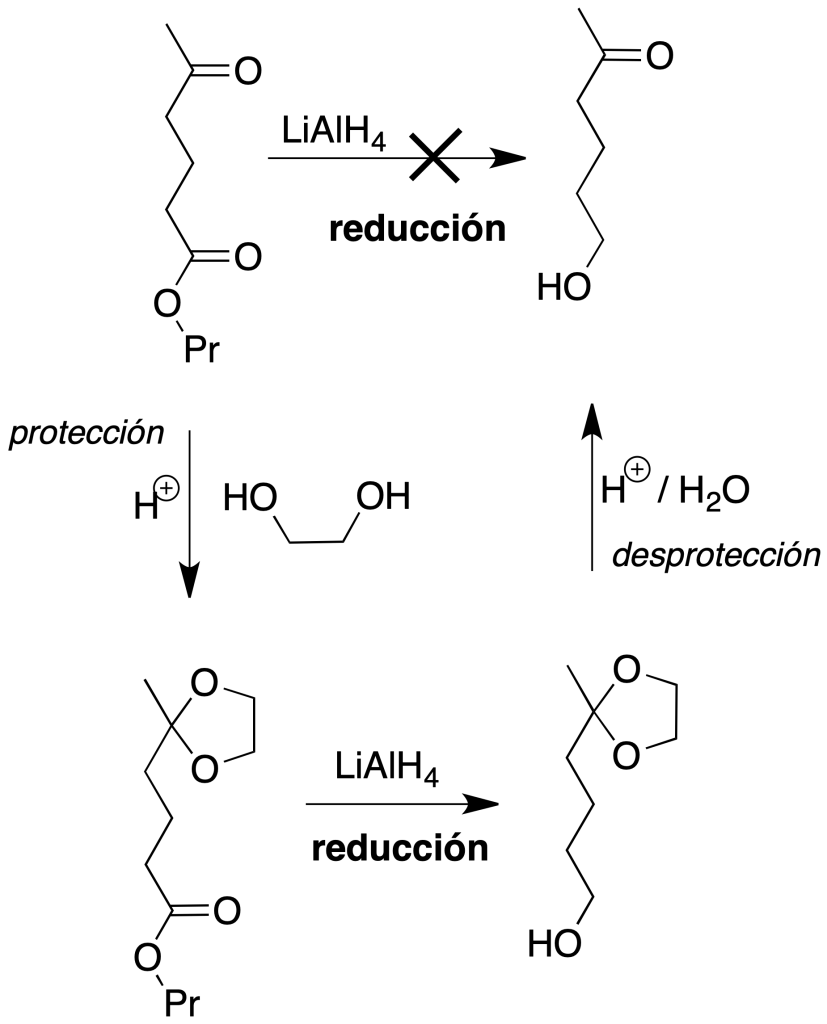
Reducción sólo del grupo éster: el tercer caso que se plantea es más complejo, ya que cualquier reductor que actúe sobre el éster reduce también al grupo carbonilo de la cetona. Para llevar a cabo la reducción del grupo éster, habrá que proteger el grupo carbonilo de la cetona previamente. Es necesario formar un derivado en el grupo carbonílico, realizar la reacción de reducción del éster, y finalmente volver a regenerar la cetona (en total, tres reacciones). Como es sabido, se denomina grupo protector al conjunto de átomos que ejerce ese papel. Los grupos protectores deben presentar las siguientes características:
- Métodos sencillos para su preparación y con buenos rendimientos.
- Compatibles con las condiciones de reacción en las que se utilizan.
- Fácil eliminación y buenos rendimientos.
Para el ejemplo elegido, el procedimiento a seguir sería el que se detalla en la figura:
Formación de enlaces carbono-carbono
Uno de los grupos de reacciones más útiles en síntesis orgánica son las de formación de enlaces carbono-carbono sencillos (C-C) y dobles (C=C), ya que permiten la construcción de esqueletos carbonados de mayor número de átomos a partir de moléculas más sencillas. Por ejemplo, las reacciones más significativas en síntesis orgánica se listan a continuación:
- Polimerización de alquenos.
- Reacción de Diels-Alder.
- Sustitución de halógeno, en haluros de alquilo, por -CN o iones acetiluro.
- Síntesis de Wurtz y Corey-House.
- Alquilación y acilación Friedel-Crafts.
- Reacción de Sandmmeyer y de Rosenmund-von Braum.
- Adición de cianuro a aldehídos y cetonas.
- Adición de organometálicos a aldehídos y cetonas.
- Reacción de Wittig.
- Adición de reactivos organometálicos a cloruros de acilo , ésteres y nitrilos.
- Reacciones de enoles y enolatos.
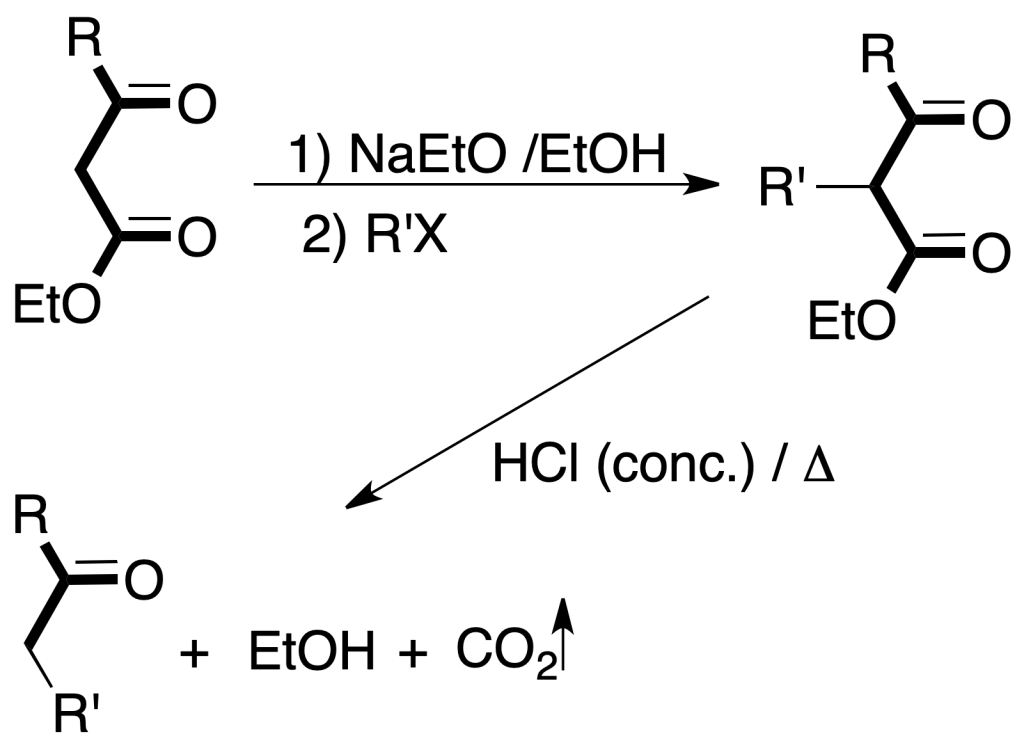
Además, son importantes los procedimientos de formación de enlaces C-C, basados en una serie de reacciones encadenadas en las que se produce la alquilación de grupos metilenos activados por sustituyentes con efecto electrón-atrayente, suelen ir acompañadas de descarboxilaciones. Por ejemplo:
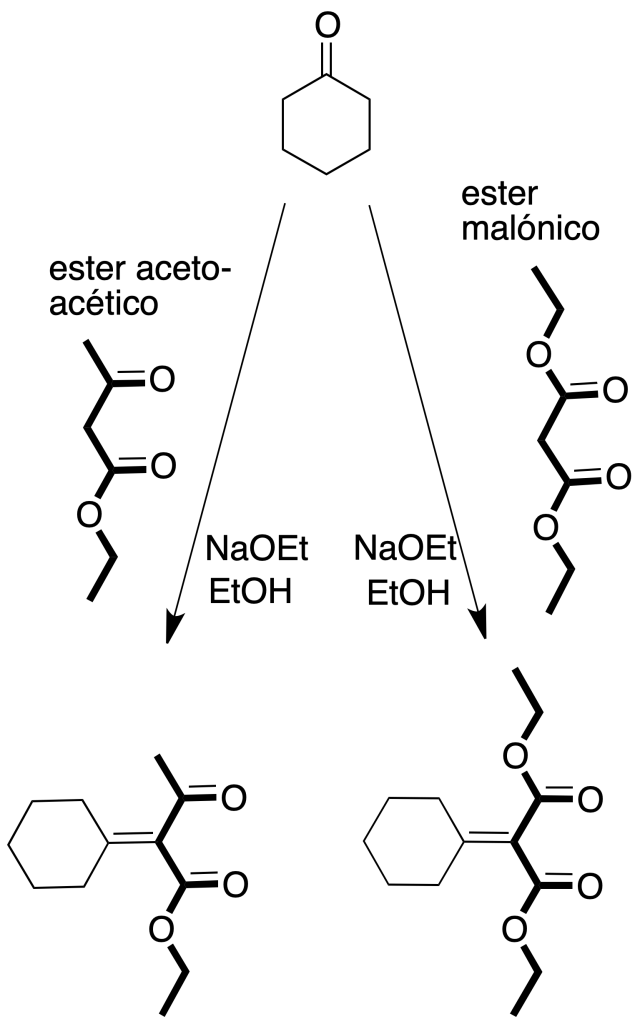
Síntesis malónica: permite la preparación derivados sustituidos del ácido acético a partir de ésteres del ácido malónico y compuestos relacionados, cuando se hacen reaccionar con un haluro de alquilo en presencia de una base.
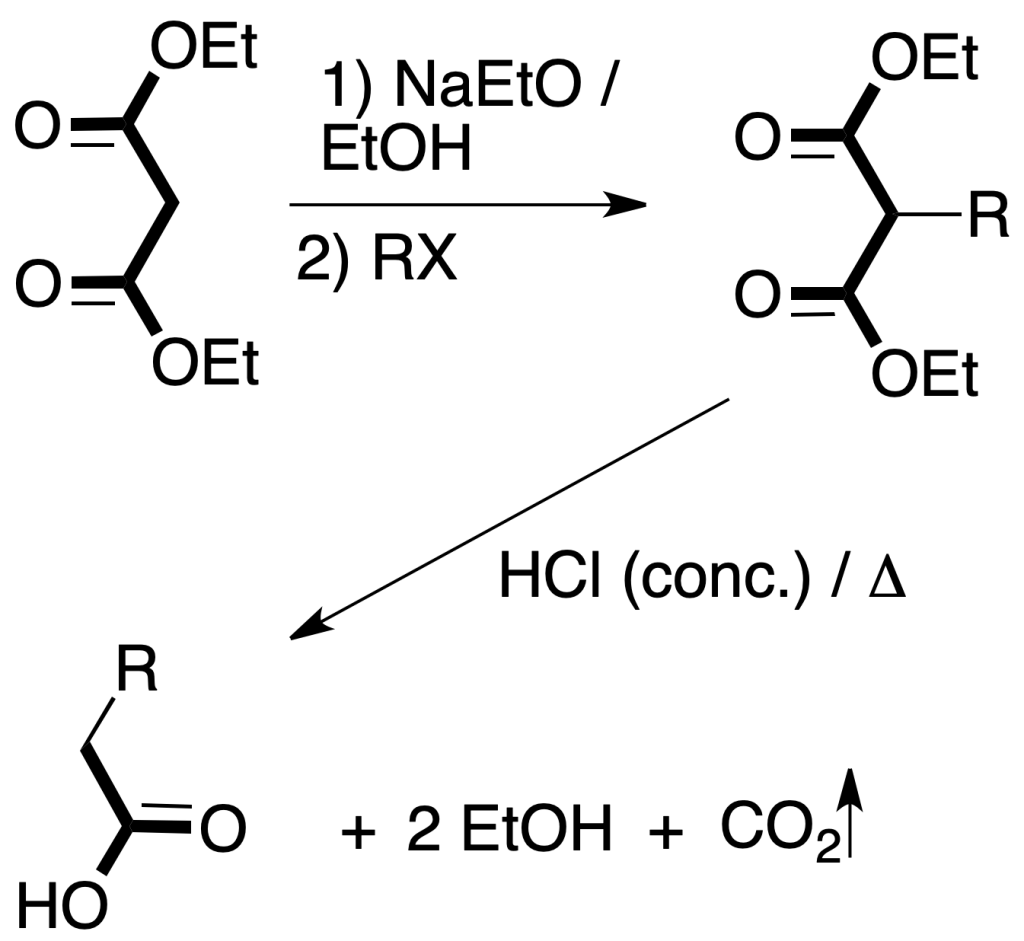
Síntesis acetoacética: se obtienen β-cetoácidos a partir de acetilacetato de etilo por tratamiento con haluro de alquilo en presencia de una base.
Condensación de Knoevenagel: se da por reacción entre un aldehído y un compuesto con un metileno unido a dos grupos electrón atrayentes (por ejemplo, β-dicarbonílico o β-dicarboxílico).
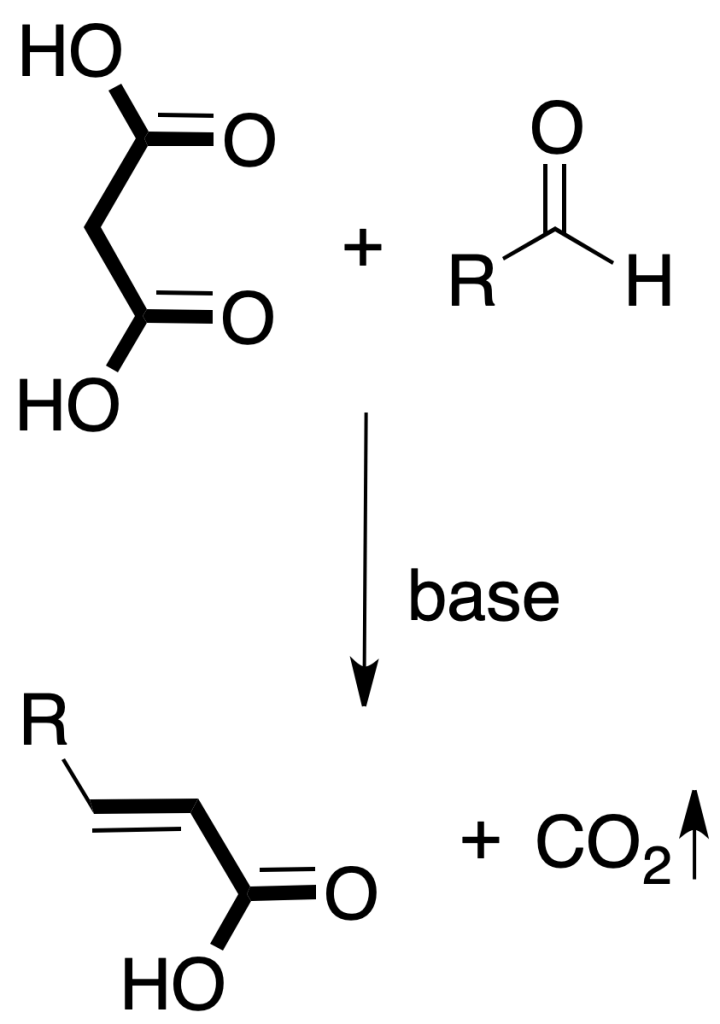
La reacción se lleva a cabo en presencia de una cantidad catalítica de base.
Tablas resumen de reactividad
Tabla 1: Reductores más usuales en Química Orgánica.
| Reactivo | Transformaciones | Disolvente / Incompatibilidad |
|---|---|---|
NaBH4 | Aldehído → alcohol 1º Cetonas → alcohol 2º Azida → amina Generalmente no reduce ácido carboxílico, amida y éster | Etanol, etanol acuoso, diglime / ácido fuerte |
B2H6 | Ácido carboxílico → alcohol 1º Aldehído → alcohol 1º Cetona → alcohol 2º Nitrilo → amina | Éter dietílico, THF / alqueno y alquino |
DIBAH | Cloruro de acilo → aldehído Amida 3º → aldehído Aldehído → alcohol 1º (lenta) Cetona → alcohol 2º (lenta) | THF, tolueno / alcohol, amina, derivado halogenado y ácido fuerte |
H2 / cat. | Alqueno → alcano Alquino → alcano Azida, nitro derivado → amina Imina, nitrilo → amina | Alcohol, éter, hidrocarburo o ácido carboxílico |
H2 / cat. (parcialmente envenenados) | Alquino → alqueno Cloruro de acilo → aldehído | Alcohol, éter, hidrocarburo o ácido carboxílico |
LiAlH4 | Aldehído → alcohol 1º Cetonas → alcohol 2º Ácido y éster → alcohol Cloruro de acilo → alcohol Epóxido → alcohol Azida, nitrilo y amida → amina Haluro y sulfonato → alcano | Éter dietílico, THF / disolvente prótico, clorado y ácido fuerte |
Metales alcalinos (Li, Na, K) | Alquino → alqueno Sistemas π-conjugados → compuestos varios | Amoníaco líquido |
Tabla 2: Oxidantes más usuales en Química Orgánica.
| Reactivo | Transformaciones | Disolvente / Incompatibilidad |
|---|---|---|
HIO4 | Diol vecinal → compuesto carbonílico | Agua pura o mezclas con otros disolventes |
MnO2 | Alcohol alílico y bencílico → compuesto carbonílico | CH2Cl2 / amina |
PCC | Alcohol 1º → aldehído Alcohol 2º → cetona | CH2Cl2 |
O3 | Rotura oxidativa de alqueno y alquino | CH2Cl2 o CHCl3 / derivados de benceno, sulfuro y amina |
RCO3H | Alqueno → epóxido Cetona → éster | CH2Cl2 y éter / amina y sulfuro |
KMnO4 | Aldehído → ácido carboxílico Alcohol 2º → cetona Alqueno → diol vecinal Alquino → ácido carboxílico | Agua pura o mezclas con otros disolvente / amina y sulfuro |
Reactivo de Collins CrO3 • 2 C5H5N | Alcohol 1º → aldehído Alcohol 2º → cetona | CH2Cl2 |
Reactivo de Jones CrO3 • H2SO4 | Alcohol 1º → ácido carboxílico Aldehído → ácido carboxílico Alcohol 2º → cetona | H2SO4 acuoso con acetona / compuestos sensibles a ácidos |
Reactivo de Swern (COCl)2 / DMSO | Alcohol 1º → aldehído Alcohol 2º → cetona | CH2Cl2 , éter o DMSO |
Pb(AcO)4 | Diol vecinal → compuesto carbonílico | Benceno o ácido acético |
OsO4 | Alqueno → diol vecinal A veces se emplea en cantidad catalítica acompañado de H2O2 | Piridina |
La síntesis orgánica en la actualidad
La búsqueda de compuestos con efectos deseados, con nuevas funciones y propiedades y el estudio de los métodos óptimos para su síntesis son ahora los objetivos principales del químico orgánico sintético. Gran parte de las síntesis orgánicas en la actualidad tiene como objetivo la preparación de compuestos con actividad biológica.
La síntesis del taxol, de las eritromicinas A y B epotilonas A y B los ácidos zaragócicos, la ecteinascidinas, las cefalostatinas o la vancomicina son algunas ejemplos de síntesis sobre las que se ha trabajado durante años.
Junto a estas síntesis, ha habido también un desarrollo espectacular en la síntesis de oligosacáridos, de peptidos y de compuestos que participan en la química de micelas. Todos ellos son importantes por su participación en diversos procesos biológicos. En los últimos años se ha realizado progresos en la síntesis de oligosacáridos habiéndose desarrollado nuevos métodos de glicosidación con eficientes grupos salientes que han dado buenos rendimientos y alta estereoselectividad bajo condiciones suaves. Las nuevas estrategias basadas en reacciones de glicosidación ortogonal son ejemplos recientes. Los péptidos como segmentos de proteínas pueden ser reconocidos por enzimas o por el sistema inmune y por esta razón, se han preparado muchos péptidos bioactivos para investigaciones farmacológicas y para estudios clínicos. Sin embargo el uso de péptidos como fármacos esta limitado debido fundamentalmente a su poca estabilidad metabólica y a problemas de transporte y asimilación. Para remediar esta desventaja y obtener compuestos útiles farmacológicamente se han desarrollado los péptidos modificados y los péptido- miméticos, algunos de ellos son prometedores agentes anticancerígenos. Estos péptidos pueden intervenir en procesos bioquímicos y biológicos complejos. Es de esperar que con el progreso en el conocimiento de los procesos celulares sea posible intervenir o inhibir selectivamente estos procesos usando péptidos modificados y péptidos- miméticos. Estos péptidos pueden ser obtenidos por diseño de su estructura o por aproximaciones de química combinatoria.
Otro campo de la Síntesis Orgánica lo constituye la síntesis de moléculas de tamaño medio y en la que dos o más grupos hidrofílicos (ácidos, aminoácidos, alcoholes, polioles, aminas y amidas entre otros) están unidos por una porción hidrofóbica (largas cadenas hidrocarbonadas, apenas ramificadas y pudiendo incluir alguna insaturación). Estos compuestos conocidos en ingles como (bolaphiles) han mostrado ser de gran interés en la química de las micelas, y su potencial en la preparación de membranas monocapas ultrafinas y en su destrucción.
Las bolaphiles como macromoléculas dendríticas también han presentado interés por su capacidad de mimetizar las enzimas, por lo que pueden actuar como agentes terapéuticos o herramientas para investigar reconocimiento y ensamblaje molecular.
Es fácil comprender que ante el gran desarrollo de la Síntesis Orgánica, es muy difícil resumir. A la vista del artículo de D. Seebach titulado «Organic Synthesis-where now?», donde tras analizar los logros más importantes en el campo de la síntesis orgánica durante los últimos años anunció “el centro primario de atención de todos los modelos sintéticos continuará desplazándose hacia las variantes catalíticas y enantioselectivas; no tardará mucho tiempo que estas modificaciones sean asequibles con todas las reacciones estándar para convertir productos aquirales en quirales”. Según esto los nuevos métodos sintéticos surgirán en los campos de la síntesis enenatioselectiva y de la Química Organometálica con especial énfasis en los métodos catalíticos.
Química Organometálica
Aunque de hecho existe una división práctica entre lo que es Síntesis Orgánica y Síntesis Inorgánica, tal división, seguramente no resistiría una argumentación conceptual medianamente rigurosa. En los comienzos del Siglo XX, el interés de los químicos orgánicos se centró únicamente en la generación de enlaces carbono carbono y en la utilización de moléculas formadas únicamente por carbono hidrogéno oxígeno azufre nitrógeno y algún halógeno. Los metales alcalinos y alcalinoterreos únicamente actuaban como contraiones en sales y alcoholatos. A esta época pertenecen muchas de las reacciones de aplicación industrial y de interés preparativo como: las condensaciones de Claisen, aldolica, la de Perkin, la reacción de diazotación de Gries, la adición conjugada de Michael, etc.
Las apariciones posteriores de la síntesis de Friedel-Craft y la de los reactivos organomagnesianos de Grignard, por una parte y por otra la hidrogenación heterogénea de alquenos sobre catalizadores de niquel, aceleraron el desarrollo de una Química Organometálica que podía complementar los métodos de síntesis de los químicos orgánicos. No obstante, su evolución fue lenta hasta que los metales de transición hicieron acto de presencia, como en el aislamiento en 1951 del ferroceno por P. Pauson y T. Kealy. El enlace carbono metal adquiría su protagonismo y no lo ha abandonado hasta hoy en día en que prácticamente ningún metal está vetado en la Química Organometálica.
Sin embargo, a pesar de las brillantes contribuciones a la Síntesis Química provenientes de los grupos principales de la tabla periódica, por ejemplo la hidroboración, la reacción sila-aldolica o las reducciones radicalarias de hidruros de estaño, parece que el protagonismo del que hicieron gala les ha sido arrebatado por los metales de transición. La naturaleza intrínsecamente diferente de los enlaces que forman los metales de transición con los ligandos orgánicos (regla de los 18 electrones, coordinación capto-dativa, etc) y los mecanismos también diferentes que operan en dichas moléculas (inserción oxidativa, eliminación reductiva, ciclos catalíticos con expansión y contracción sucesiva de la esfera electrónica del metal, etc.) hacen que los ligandos orgánicos unidos a metales de transición puedan sufrir, por ejemplo, reacciones de sustitución en átomos de carbono sp2 vinílicos o aromáticos y otras muchas transformaciones interesantes, difíciles de conseguir con los métodos de la Química Orgánica tradicional.
La capacidad demostrada de los metales de transición para generar ciclos catalíticos en los que se forman enlaces C-C, ha sido la desencadenente de la denominada metalo-catálisis en sus diferentes formas: homogénea, hetereogénea y homogénea-heterogeneizada. El auge que ha tenido, ha sido tal que hoy sería imposible prescindir de la catálisis tanto en la petroquímica, como en cualquier industria que manufacture compuestos orgánicos. Por ello, es conveniente hacer algunas consideraciones sobre el binomio metalo-catálisis/Síntesis Orgánica que luego puede tener importancia en la docencia de ambas, enfatizando las ventajas que presentan los procesos metalo-catalizados, a saber: brevedad, alta quimio-, regio- y en algunos casos estereo-selectividad, baja o nula generación de subproductos, condiciones de reacción suaves y alta productividad.
Otro aspecto muy importante de los compuestos organometálicos es la denominada química organometálica de superfícies que está intimamente relacionada con la denominada catálisis homogenea hetereogeneizada, y se refiere al estudio de las reacciones de los complejos organometálicos con la superficie de ciertos compuestos sólidos como óxidos metálicos, ceolitas y metales. Se trata, en realidad, de un nuevo aspecto de la Química de la Coordinación de los compuestos organometálicos que puede rivalizar, con la catálisis realizada por los compuestos organometálicos tanto en fase homogénea como en condiciones hetereogéneas. Esta aproximación es la denominada Química Organometálica de superficies. Se ha comentado que la Química Organometálica Sintética puede ser la base para desarrollar la síntesis de catalizadores a la carta en el sentido de que puede permitir la preparación meticulosa del centro activo. Sin embargo, y aunque ciertamente existen aplicaciones industriales de la técnica, parece que una de sus limitaciones puede ser el escalado de las reacciones.
Síntesis asimétrica
Desde los descubrimientos de L. Pasteur hasta la actualidad, puede afirmarse que la preparación de compuestos enantioméricamente puros ha experimentado tal auge durante los últimos 15 años que constituye en este momento el área de la síntesis orgánica que más publicaciones genera. Paralelamente, también se ha creado un importante enriquecimiento conceptual, donde tienen cabida conceptos como inducción asimétrica múltiple, amplificación de quiralidad, catálisis acelerada por ligandos o la autoregeneración de estereocentros etc. Con todo esto, las tendencias generales que se perfilan como importantes en la síntesis asimétrica son:
- La preparación de compuestos enantioméricamente puros mediante síntesis asimétrica estequiométrica. Mucho trabajo se hizo en la década de los ochenta para el desarrollo de auxiliares quirales eficaces para estos procesos. Aunque las contribuciones son importantes, el hecho de que requieran dos etapas de síntesis adicionales (el anclaje del auxiliar quiral a su sustrato y su posterior eliminación del producto) hacen girar la vista hasta los procesos catalíticos.
- La preparación de productos enantioméricamente puros mediante biocatálisis. Algunas contribuciones a la síntesis asimétrica biocatalizada han aparecido utilizando tanto enzimas como organismos vivos, demostrando en ambos casos la capacidad catalítica de los enzimas tanto a nivel industrial como de laboratorio. Dentro de esta tendencia, hay que citar la catálisis anticuerpo que se utiliza con gran éxito en procesos tales como: esterificación e hidrólisis de esteres, ruptura de uniones peptídicas, lactonizaciones, procesos redox, transposiciones de Claisen, reacciones de Diels-Alder, etc. Es de esperar que en un futuro próximo investigaciones de este tipo utilizando análogos de estados de transición puedan competir con la denominada metodología del punto de mutación en los estudios sobre proteinas.
- La preparación de compuestos enantioméricamente puros mediante síntesis enantioselectiva catalítica, enzimas artificiales. Estos catalizadores son capaces de realizar las mismas funciones que los enzimas con la ventaja de que pueden trabajar en condiciones mas flexibles y que su estructura puede ser modificada para influir en su comportamiento.
El uso de los ordenadores en síntesis orgánica
El análisis del proceso mental seguido para concebir la síntesis de una molécula orgánica compleja puede resultar tan complicado como la propia síntesis. De todos es conocido tal como hemos puesto anteriormente de manifiesto el ya clásico debate mantenido durante los años 70 por los profesores R.B. Woodward y E.J. Corey, en torno a si la síntesis es un arte dependiente de la genialidad y de la experiencia del químico o una actividad lógica, estructurable y sistematizable, aún en los casos mas difíciles y complejos. Las consecuencias que para los docentes pueden tener estos dos enfoques son muy importantes, ya que el proceso de creación artística no se puede transmitir al estudiante y el de una actividad 100% racional si. Con el paso del tiempo, es la segunda opción la que se va consolidando y hoy en día los conceptos de sinton, retron, desconexión, etc. son incluidos sistemáticamente en los contenidos de los cursos de Síntesis Orgánica.
Como exponente más sofisticado de la tendencia anteriormente mencionada, la práctica de la investigación en Síntesis Orgánica cuenta con programas de ordenador tales como “CAMEO” (Computer Assisted Mechanistic Evaluation of Organic Reactions), “CASP” (Computer Asssisted Synthesis Plan), “ORAC” (Organic Reaction Access by Computer), “SYNLIB” (Synthetic Library) o “CHAOS” (Computerization and Heuristic Applied to Organic Synthesis) etc. Que combinan la asequibilidad y la facilidad de manejo con altos grados de fiabilidad. Aunque en este momento obsoleto, no se puede dejar de mencionar el primigenio “LHASA” (Logic and Heuristic Applayed to Synthetic Analysis) ideado por Corey en los años 70.
Desarrollos actuales en Síntesis Orgánica
La Síntesis Orgánica es una de las parcelas de la Química Orgánica que más ha evolucionado; el mayor conocimiento de la Química Teórica, de los mecanismos de reacción por un lado y el desarrollo de los métodos de aislamiento purificación y determinación estructural por otro han contribuido a ello; de aquí que el avance más espectacular de la Síntesis Orgánica comience a partir de los años 50. Un punto de referencia en el campo de la Síntesis Orgánica, lo constituye la síntesis de la estricnina descrita por R.B. Woodward en 1954, quien recibió el premio Nobel en 1965 por sus logros en el campo de la Síntesis Orgánica.
La síntesis de la estricnina por Woodward puede considerarse un hito en el desarrollo de la moderna Síntesis Orgánica, tanto por el diseño como por la ejecución de la síntesis de una molécula tan compleja. La segunda síntesis total de la estricnina fue realizada por P. Magnus en 1992. En 1993, L.E. Overman publicó la primera y hasta el momento la única síntesis enantioselectiva.
Otro hito en la Síntesis Orgánica lo marcó E.J. Corey (Premio Nobel en 1990), quién aplicó de forma sistemática nuevos principios y metodologías en el diseño de síntesis. Como el mismo analiza, durante la primera mitad del siglo XX la Química Orgánica centraba su atención en conocer su estructura carbonada y las transformaciones de los compuestos orgánicos. Los cambios químicos se miraban en la dirección de la reacción química, es decir desde reactivos a productos. La mayoría de las síntesis desarrolladas se llevaron a cabo seleccionando el material de partida apropiado e investigando una serie de reacciones que transformaran el material en el producto deseado. A mitad de los años 60 se desarrolla una aproximación diferente y más sistemática, que consiste en analizar estructuralmente el producto de reacción y en la manipulación de la estructura en sentido inverso. Esto es lo que se conoce como análisis retrosintético y fue desarrollado por E.J. Corey. Este autor fue el precursor del diseño actual de síntesis por ordenadores. Estos dos padres de la Síntesis Orgánica, Woodward y Corey, la conciben como arte o como ciencia. El profesor F. Serratosa la resume de la manera siguiente: “para R.B. Woodward, la síntesis orgánica es fuente de emoción, provocación y aventura y puede ser también un arte noble; para E.J. Corey la Síntesis Orgánica es una actividad esencialmente lógica y racional”. El mismo E.J. Corey así lo indicaba en su libro “ The logic of Chemical Synthesis” .
A partir de estos autores la Síntesis Orgánica ha experimentado un avance tan espectacular que resulta imposible enumerar las síntesis descritas y los investigadores que han contribuido a ello. El auge de la síntesis orgánica a partir de los años 60 ha sido tan grande que antes de comenzar la década de los 90 han sido sintetizados miles de compuestos con estructuras complejas. La complejidad de los sistemas sintetizados parece culminar con la síntesis de la palitoxina por Y. Kishi en 1987, la toxina natural más potente que se conoce con 64 estereocentros y 7 dobles enlaces para los que son posibles 271 estereoisómeros.
Los grandes avances que ha experimentado la Síntesis Orgánica ha hecho preguntarse a algunos autores si como ciencia ha alcanzado sus límites, ya que hoy en día puede considerarse que la síntesis de cualquier estructura, por compleja que sea, puede realizarse si se tiene acceso a los conocimientos, se usa la imaginación y se dispone de soporte económico suficiente. Por otro lado alguno de los objetivos clásicos que se aborda en la síntesis de un compuesto, tales como la confirmación de la estructura de un producto natural, el interés teórico o el reto intelectual, por conseguir una síntesis total, parece que han perdido cierta vigencia.
Listado de ejercicios resueltos de síntesis orgánica
Siga el enlace para ver un listado de problemas resueltos sobre síntesis orgánica.
