Índice
Introducción
Los heterociclos no aromáticos pueden relacionarse con los compuestos análogos de cadena abierta, pero el tamaño del anillo puede jugar un papel relevante entre ellos. La existencia de un sistema anular impone restricciones a la molécula que pueden estar ausentes en el sistema acíclico.
Cuanto más grandes sean estas restricciones mayor será la diferencia entre ambos sistemas. Las moléculas flexibles adoptan, preferentemente, conformaciones en las que las interacciones de enlace se maximizan y se minimizan las interacciones repulsivas no enlazantes.
En estas conformaciones los ángulos y las distancias de enlace serán los «naturales». Si la presencia de un anillo obliga a la molécula a adoptar una estructura en la que no sea posible lograr estas características prefereidas, se puede considerar que la molecula está «tensionada».
Aun así, esta va a adoptar preferentemente, una forma que maximize las interacciones atractivas o de enlace, y minimize las repulsivas.
Las tensiones anulares en los heterociclos no aromáticos se pueden considerar como la suma de una serie de factores:
- Tensión angular de los enlaces.
- Tensión o compresión de enlace.
- Torsión de enlace.
- Interacciones no enlazantes.
De alguna manera todos estos factores están interrelacionados y combinando cualquiera de ellos se modifican los otros.
Tensión angular de los enlaces
A menudo se presentan distorsiones en los ángulos «naturales» de los sistemas cíclicos. La energía Vα requerida para distorsionar el ángulo de enlace, respecto a su valor de equilibrio, se conoce como «tensión de ángulo de enlace» o «tensión de Baeyer«. Viene dada por la siguiente expresión:
Vα = 0.01 (α-α0)2
Donde α es el valor real del ángulo y α0 el valor natural del ángulo. Una distorsión de 10º lleva asociada una contribución energética de 1 kcal/mol, 20º le corresponde 4 kcal/mol, y a 50º es de 25 kcal/mol.
Tensión angular en heterociclos pequeños
En los anillos de 3 miembros se encuentran las mayores distorsiones. Los anillos saturados de 3 miembros presentan ángulos de enlace de 60º, que se se comparan con los aproximadamente 110º de un carbono con hibridación sp3, le corresponde una gran distorsión. Llegando incluso a ser mayor para los anillos insaturados.
En la siguiente Tabla 1, se muestra la tensión correspondiente a los anillos saturados de 3 miembros (calculada en base a las entalpias de formación experimentales y estimadas).
| ∠CXC (º) | C—C (Å) | C—X (Å) | Tensión (kcal/mol) | |
| CH2 | 60 | 1.510 | 1.510 (1.54)* | 27.5 |
| NH | 60 | 1.481 | 1.475 (1.47) | 27.1 |
| CH3—SH | 48.5 | 1.492 | 1.819 (1.81) | 19.9 |
| CH3—OH | 61 | 1.472 | 1.436 (1.43) | 27.2 |
| *Longitudes promedio del sistema no tensionado (sp3-sp3). | ||||
Los efectos de la distorsión de los ángulos de enlace se minimizan en estos compuestos de anillo pequeño debido a cambios en la hibridación de los átomos que forman los anillos.
Por ejemplo, en los anillos saturados de tres miembros, los enlaces que forman el anillo no están constituidos por híbridos sp3, sino que tienen más carácter «p», permitiendo un solapamiento más efectivo de los orbitales dirigidos hacia fuera de los ejes que unen a los núcleos de los átomos del anillo.
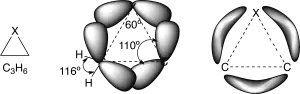
El resultado es que las distorsión inter-orbital es mucho menor que la distorsión angular inter-nuclear. Porque, conforme se incrementa el carácter «p» de los enlaces del anillo, el ángulo inter-orbital «natural» disminuye del valor de 109.3º (sp3) -> 90º (p ortogonales).
Se pueden considerar a los átomos del anillo como unidos por enlaces doblados o en forma de banana, (enlace entre σ y π) con una densidad de carga baja y dirigida hacia fuera del anillo.
Este cambio en los enlaces del anillo, afecta también a otros enlaces, los que los unen a los sustituyentes. Estos poseen un carácter «s» mucho mayor de lo normal y los ángulos son mas grandes.
Consecuencias de la tensión angular en anillos pequeños
- Gran reactividad y particularmente fácil apertura.
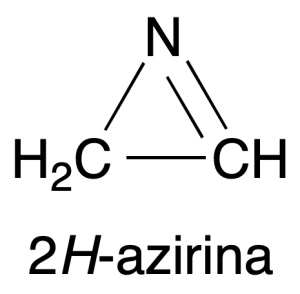
- Diferentes propiedades espectroscópicas. Por ejemplo la frecuencia de tensión del enlace C=N, en el espectro de Infrarrojo (IR) de la molécula 2H-azirina, ν(C=N), aumenta hasta 1800 cm-1 en comparación on el valor normal de 1650 cm-1 para iminas no tensionadas.
- La distancia entre elementos en oxiranos y aziridinas es más corta de lo normal (transición conjugación), con más carácter p, doble enlace parcial y solapamiento π-orbital-p-anillo.
- El mayor caracter «s» del par de orbitales del nitrógeno en aziridinas, con respecto a las aminas, hacen que sea menos básico que las aminas alifáticas secundarias (con pKa = 8.04). Además, el par solitario intercatúa con menos eficiencia que en otras aminas con sustituyentes conjugativos, como el fenilo unido al nitrógeno (orden de basicidad: heterociclos de 4 miembros > heterociclos de 3 miembros).
- En las aziridinas se reduce considerablemente la velocidad de inversión piramidal del nitrógeno (aumenta la barrera) en comparación con otros compuestos de este elemento:
fig-01
- Las aminas sencillas no tensionadas presentan barreras bajas de inversión del nitrógeno (ΔG‡ ≅ 6 kcal/mol), siendo la barrera en la aziridina (ΔG‡ ≅ 17 kcal/mol) mucho mayor.
Esto se debe a la mayor tensión angular en el estado de transición plano que se requiere para la inversión. Hay mayor tensión angular en el estado de transición y la barrera de inversión es mayor.
- La barrera de inversión se ve influenciada, también, por la naturaleza del sustituyente del nitrógeno.
Los sustituyentes electrón atrayentes, que estabilizan el estado de transición plano, reducen la barrera energética.
Puede existir también un efecto estérico que desestabiliza a las formas piramidales por ejemplo el grupo terbutilo (tBut) disminuye la barrera.
Tensión angular en anillos grandes
Si nos fijamos en la molécula de 1-azabiciclo[3.3.3]undecano, podemos observar que presenta una disposición casi planar de los grupos sobre el nitrógeno.
fig-02
Aquí, el efecto es opuesto al observado en anillos pequeños. Los ángulos son ahora mayores que los naturales. Además, a medida que el anillo aumenta de tamaño, este efecto se hace más notable. El compuesto adopta preferentemente una conformación en la que el par de electrones sin compartir del nitrógeno se dirige hacia el interior de la jaula.
Como resultado, la molécula presenta una gran dificultad a protonarse en el nitrógeno por la cara exterior, resultando en una base muy débil.
Barreras energéticas de torsión
La torsión de enlace hace referencia a la rotación alrededor del eje que une a dos núcleos. El ángulo de torsión o ángulo dihedro es la medida de la rotación del enlace respecto a la posición en la que los sustituyentes del núcleo están eclipsados.
En los sistemas flexibles, la energía de la molécula varía conforme gira el enlace.
Ejemplos
- En la molécula de etano CH3-CH3, la diferencia entre la forma de mayor energía (eclipsada) y la de menor energía (alternada) es de aproximadamente 2.9 kcal/mol.
- La barrera energética para las dos formas silla del ciclohexano es aproximadamente 10.5 kcal/mol.
La naturaleza química de dicha barrera es compleja y puede contener contribuciones de la interacción de núcleos y de los pares de electrones sin compartir de heteroátomos.
Enlaces sencillos
Los heterociclos saturados contienen enlaces C—X y en algunos casos X—X, como parte del sistema anular, para los que las barreras de rotación son distintas de las correspondientes a los enlaces C—C.
Los enlaces sencillos C—X, presentan, en general, barreras de rotación más bajas que las de los enlaces C—C.
| Molécula | Energía (kcal/mol) |
| CH3—CH3 | 2.9 |
| CH3—NH2 | 2.0 |
| CH3—SH | 1.3 |
| CH3—OH | 1.1 |
Por otra parte, los enlaces X—X, presentan unas barreras de rotación mucho más altas que los enlaces C—C.
Ejemplo de hidrazina
La hidrazina (NH2—NH2) presenta una barrera de rotación de 11.9 kcal/mol. Esto se debe, principalmente, al par de electrones sin compartir de los nitrógenos.
Si consideramos el perfil de energía rotacional de la hidrazina.
fig-03
El rotámero que posee mayor energía es aquel que los pares de electrones sin compartir están eclipsados y los mínimos cuando los pares de electrones están a 90º.
La barrera de rotación es menor para el confórmero en el que los pares de electrones sin compartir están en disposición trans (uno respecto al otro).
Efecto gauche
Los heterociclos que contienen enlaces, muestran preferencias por conformaciones en las que los orbitales con pares de electrones sin compartir se encuentran lo más perpendiculares posibles.
Un compuesto será menos estable si se halla restringido a conformaciones en las que los pares de electrones sin compartir se encuentran eclipsados.
Ejemplo de diaziridinona
- En la siguiente molécula monocíclica, los sustituyentes y los pares de electrones están en conformaciones trans, por tanto dicha molécula es estable.
fig-04
- En la siguiente molécula bicíclica, los sustituyentes y pares de electrones se encuentran eclipsados, y por tanto dicha molécula es iniestable. Pierde rápidamente el monóxido de carbono (CO) a una temperatura de 25ºC, y se rompe con facilidad al reaccionar con nucleófilos.
fig-05
Enlaces dobles y enlaces dobles parciales
Existen heterociclos que incorporan en su estructura enlaces π. La regla de Bredt, formulada hace muchos años, prohibe la formación de dobles enlaces sobre cabezas de puente. Posteriormente, a su formulación, se reconoció que el criterio de tensión en dichos compuestos venía impuesto por el tamaño del anillo más pequeño que presentaba la olefina trans, y si este es lo suficientemente grande, podría existir (ciclos de 8 o más miembros).
Así pues, se conocen unos cuantos heterociclos con enlaces C=N en cabeza de puente, en los que los grupos unidos al enlace π no pueden ser coplanares.
Como consecuencia estos heterociclos son muy reactivos como se ilustra en la siguiente figura.
fig-05
Preferencias conformacionales de los heterociclos saturados
Hemos visto que la energía torsional del enlace C—X es menor que la del enlace C—C, mientras que la del enlace X—X es más alta.
Estas diferencias son muy impactantes en los heterociclos flexibles, aunque hay otros factores que también influyen en la conformación preferida:
- Diferencia de las longitudes de enlace C—X comparados con los enlaces C—C.
- Diferencia de los radios de Van der Waals.
- Presencia de electrones sobre los heterociclos.
Los enlaces C—O presentan unos valores de 1.43 Å, y los C—N valores de 1.47 Å, mucho más cortos que los de los enlaces C—C con valores de 1.51 Å.
Los radios de Van der Waals, también aumentan en el orden: O < NH < CH2, con valores de 1.4 < 1.5 < 2.0 Å, respectivamente.
Heterociclos saturados de 6 miembros
De la misma manera que el ciclohexano y derivados adoptan la conformación silla, también lo hacen los heterociclos de 6 miembros.
La barrera de energía conformacional de inversión del anillo en el tetrahidropirano es de 9.9 kcal/mol. Este valor es muy similar al del ciclohexano, como lo es también para otros heterocilos de 6 miembros.
fig-06
Si tenemos en cuenta el heterociclo sustituido 3-metil-1,3-dioxano:
fig-07
la conformación A, con el metilo ecuatorial, se ve aún más favorecida que en el metilciclohexano (1.8 kcal/mol).
fig-08
Esto se debe a que los enlaces más cortos C—O generan interacciones 1,3-diaxiales mayores en B que en el metilciclohexano.
Por otra parte, en el caso del 5-metil-1,3-dioxano de la figura:
fig-09
La conformación A, es más estable pero sólo en 0.8 kcal/mol. Esto se debe a que en la conformación B la interacciones del grupo metilo con los pares de electrones sin compartir del oxígeno son mucho más pequeñas que las interacciones 1,3-diaxiales con los enlaces C—H presentes en el metilciclohexano.
La piperidina y otros heterociclos nitrogenados sufren un conjunto más complicado de cambios de conformación. Así, tiene lugar, por una parte, la inversión del anillo y además la inversión tetraédrica de los sustituyentes del nitrógeno.
fig-10
Esta inversión tetraédrica en el nitrógeno es el proceso de menor energía.
El invertómetro en que el nitrógeno es ecuatorial es más estable. Además, los grupos alquilo unidos al nitrógeno, también, ocupan posiciones ecuatoriales y todavía más en sistemas bicíclicos.
fig-11
La conformación A es más estable que B, en 4.5 kcal/mol.
Las barreras de inversión del nitrógeno aumentan considerablemente cuando hay heteroátomos junto al heteroátomo anular. Estos pueden proceder de dentro o fuera del anillo.
fig-12
Heterociclos de 4 y 5 miembros
El ciclobutano es una molécula no plana. La barrera de energía para la inversión es baja, soló de 1.5 kcal/mol.
fig-13
Para los heterociclos de 4 miembros como la azetidina y el oxetano, existen también barreras energéticas similares, aunque más bajas.
fig-14
En el oxetano la barrera energética está por debajo del nivel basal de la energía vibracional. Por tanto, la molécula es plana como consecuencia de la baja energía de torsión del enlace C—O.
Los heterociclos de 5 miembros se pueden representar por una serie de estructuras no planas, de interconversión libre, como la estructura denominada sobre o la semisilla.
fig-15
Como en el caso de otros heterociclos, las barreras de torsión son mayores en los enlaces unidos a dos heteroátomos y se reducen las velocidades de inversión del nitrógeno tetraédrico. Por tanto, se pueden separar como diasteroisómeros las moléculas que se indican en la figura:
fig-16
Otros tipos de interacción en los heterociclos saturados
Efecto anomérico
La presencia de pares de electrones sin compartir en heteroátomos de un heterociclo, puede tener efectos sobre el sistema de enlaces en otras partes de la molécula. El efecto anomérico se debe a las interacciones de orbitales a través de enlace.
Un orbital con el par de electrones con la orientación apropiada puede mezclarse con los orbitales enlazantes y anti-enlazantes de energía similar y afectar a la fuerza de esos enlaces.
Una interacción de este tipo, es la que tiene lugar en el par de electrones y un enlace σ antiperiplanar de un átomo adyacente.
fig-17
Es posible una interacción intensa a causa de la alineación del par de electrones con el enlace σ. Se puede representar por medio de las estructuras resonantes siguientes:
fig-18
El par de electrones puede interaccionar tanto con el orbital σ como con el σ* de C—Y. Pero si (Y) es electronegativo la segunda interacción es más importante.
Los resultados son la transferencia de la densidad electrónica del par de electrones hacia σ*, fortaleciéndose el enlace C—X y debilitándose el enlace C—Y.
Si la interacción es estabilizadora, las moléculas flexibles deberán adoptar, preferentemente, conformaciones que la maximicen.
Ejemplos
En los derivados de tetrahidropiranos y otros heterociclos oxigenados y de azufre, los grupos electronegativos (F, Cl, Br, OR) en el carbono C-2 tienden a ocupar preferentemente la posición axial, aunque se encuentre más impedida estéricamente.
Se le denomina efecto anomérico porque se estudió por primera vez en la química de monosacáridos, en concreto, en los equilibrios anoméricos de los glicósidos α y β.
El efecto anomérico se ve influido por la presencia de otros sustituyentes y también por el efecto del disolvente.
fig-19
Los disolventes con fuerte tendencia a formar enlaces de hidrógeno, también estabilizan, preferentemente, los grupos alcoxi ecuatoriales.
Los efectos sobre la reactividad química son significativos debido a la interacción de los pares de electrones con los enlaces antiperiplanares.
fig-20
El conformación A es más resistente a la hidrólisis que la conformación B, que presenta el grupo ariloxi axial.
La conformación A no se afecta por el HCl 0.1 M despúes de varias semanas a 39 ºC. Sin embargo, la conformación B se hidroliza rápidamente en las mismas condiciones en 7.7 min.
Interacciones atractivas a través del espacio
El ejemplo más sencillo de estas interacciones sería el enlace de hidrógeno que puede presentarse.
Por ejemplo, entre un heteroátomo del anillo y un sustituyente hidroxilo.
fig-21
La molécula de 3-hidroxiperidina puede existir en la forma A con el hidroxilo en axial.
Otro tipo de interacción atractiva se da en aquellos compuestos que presentan heteroátomos nucleofílicos y átomos de carbono electrofílicos en las posiciones en que puedan interactuar.
Lo mismo ocurre en los compuestos de cadena abierta que poseen estas dos funciones separados por una cadena de 3 o 4 átomos de carbono que pueden formar un ciclo.
fig-22
En los ciclos ocurre lo mismo. Varias aminocetonas anulares adoptan preferentemente conformaciones que permiten al átomo de nitrógeno nucleofílico aproximarse a un átomo de carbono electrofílico en el lado opuesto del anillo.
Ejemplos
la clivorina (alcaloide) en su estructura cristalina (Rayos-X), el átomo de nitrógeno se halla anormalmente cercano al grupo carbonilo (1.99 Å) y está dirigido hacia dicho grupo en el ángulo necesario para la formación incipiente de un enlace σ.
fig-23
Las consecuencias se manifiestan en los compuestos más sencillos de este grupo, por ejemplo, la aminocetona de la figura:
fig-24
que es debilmente básica y se protona en el oxígeno más que en el nitrógeno para dar una sal de amonio bicíclica.
fig-25
Catedrático de Química Orgánica en la Universidad de Granada, con una larga trayectoria en Química Computacional, en modelado y diseño molecular.