Índice
¿Qué son las piridinas?
La piridina es un líquido incoloro que hierve a 115 ºC. Se solía obtener casi exclusivamente del alquitrán de la hulla. Hoy día, se obtiene industrialmente partiendo de amoniaco y acetileno.
La piridina al igual que el benceno tiene 6 electrones π (aromático Hückel). Su estructura es plana con ángulos de enlace de 120º. El calor de combustión revela que la energía de resonancia es de 23 kcal·mol-1.
Al igual que el benceno presenta el sexteto aromático, pero a diferencia de los heterociclos pentagonales, el par de electrones del nitrógeno no interviene en la aromaticidad. La piridina presenta propiedades básicas. La introducción de un heteroátomo en el anillo bencénico hace que sean posibles más estructuras canónicas en el hibrido de resonancia. En concreto, cinco estructuras resonantes para el caso de la piridina.
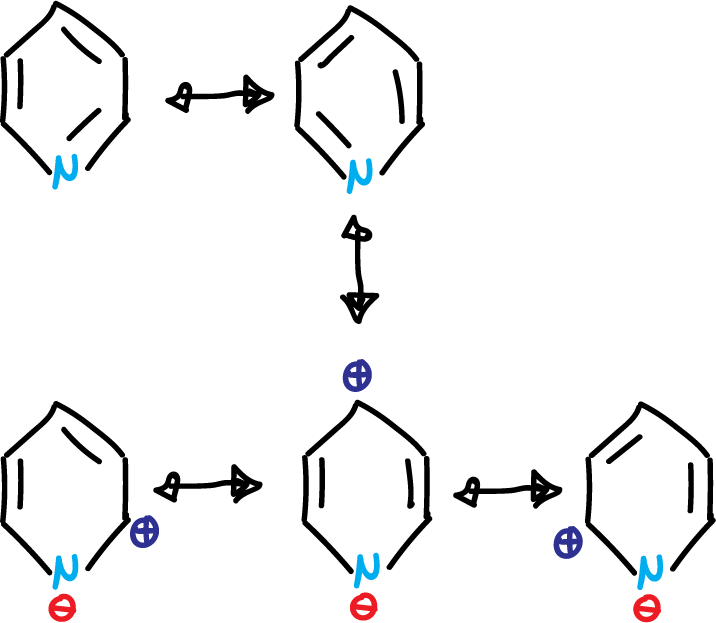
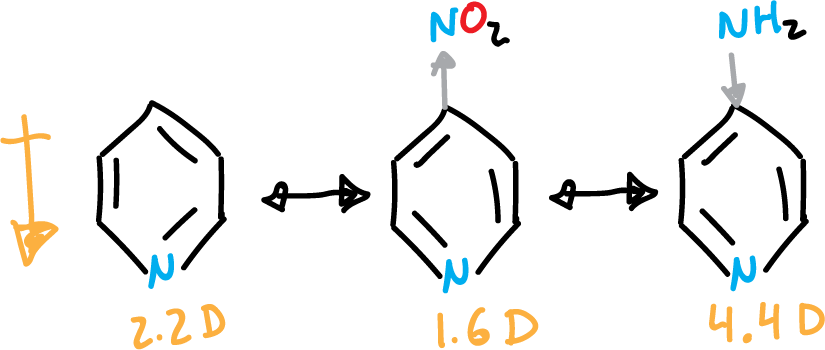
Debido a las 3 últimas resaltadas en la figura, el anillo queda con un resto de carga positiva (⊕), lo cual dificulta la sustitución electrofílica aromática (SEAr). También, hace que la piridina presente momento dipolar (μ = 2.2 Debye). La existencia de un grupo donante de electrones en posición C4 aumenta ese momento dipolar y un grupo electrón atrayente en la misma posición hace que disminuya.
Al presentar un átomo de nitrógeno con un par de electrones que no interviene en la aromaticidad. En consecuencia, las piridinas pueden protonarse para formar los ácidos conjugados correspondientes, o iones piridinio.
La fortaleza como base de las piridinas varía según la naturaleza del sustituyente del anillo. Así, los grupos donantes de electrones aumentan el valor del pKa, mientras que los grupos electrón atrayentes lo disminuyen.
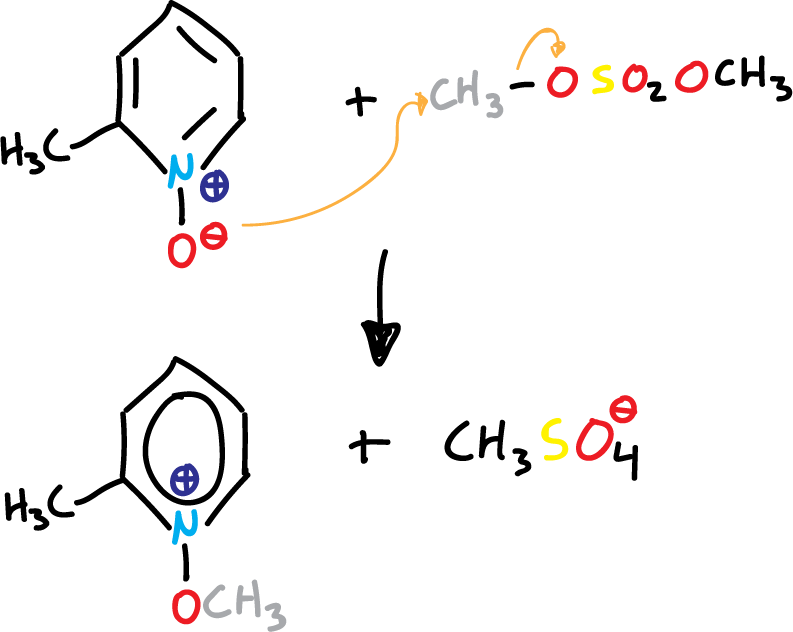
Por tanto, las piridinas pueden formar complejos con gran diversidad de ácidos de Lewis. Esto es, reaccionan con agentes alquilantes para dar sales de piridinio. También, pueden oxidarse para dar N-óxidos de piridina, mediante tratamiento con perácidos.
Los N-óxidos de piridina, desde el punto de vista estructural, constituyen unos compuestos relevantes ya que presentan la capacidad de aumentar la densidad electrónica en diversas posiciones del anillo. O a la inversa, dependiendo del reactivo frente al cual se encuentren.
Este fenómeno, se hace evidente en las estructuras que contribuyen al híbrido de resonancia del N-óxido de piridina. Al igual que en el caso de la piridina, las formas dipolares contribuyen considerablemente.
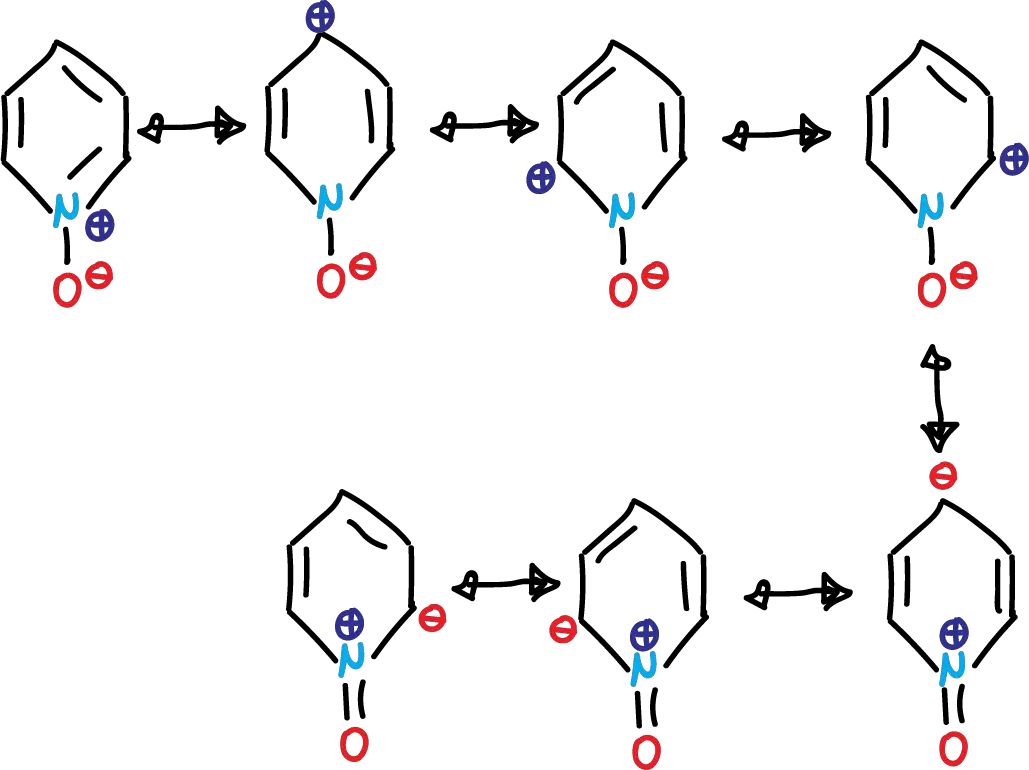
Si observamos a las 4 primeras estructuras, el átomo de oxígeno debe ser capaz de actuar como nucleófilo. Esto se evidencia en las reacciones de los N-óxidos de piridina con gran variedad de agentes alquilantes. De esta manera, el resultado es un desplazamiento de tipo SN2 para formar sales de N-alcoxipiridinio.
Síntesis de piridinas
Los métodos clásicos para obtener piridinas a partir de precursores acíclicos utilizan las reacciones de ciclación.
Así, teniendo en cuente un análisis retrosintético:
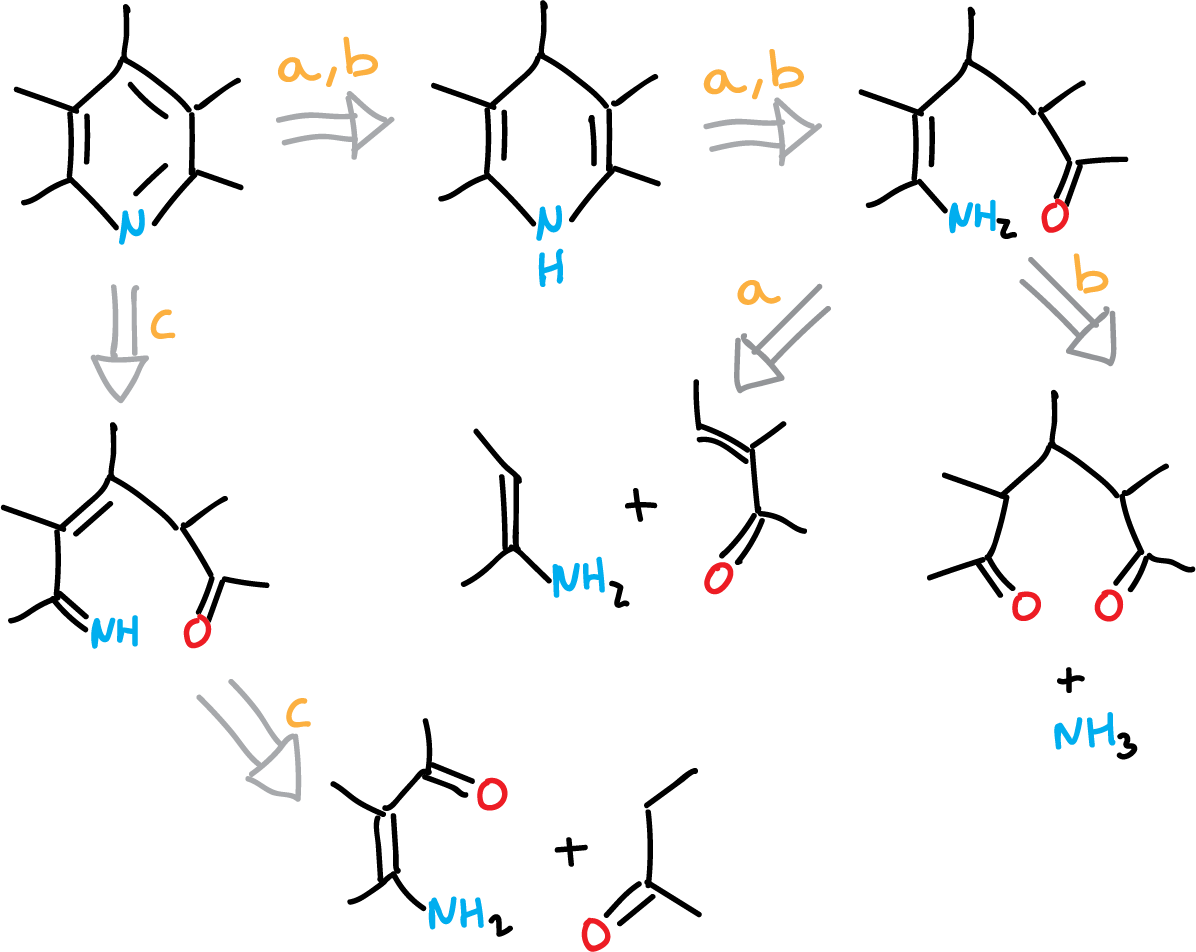
Tendríamos tres maneras de formar el anillo a partir de materias primas fácilmente disponibles.
Estos tres métodos (rutas a, b y c) junto con otros adicionales se emplean para la síntesis de piridinas.
Síntesis de Hantzsch de piridinas
Consiste en la condensación de 2 moléculas de un β-cetoéster con un aldehído y amoniaco (ruta a).
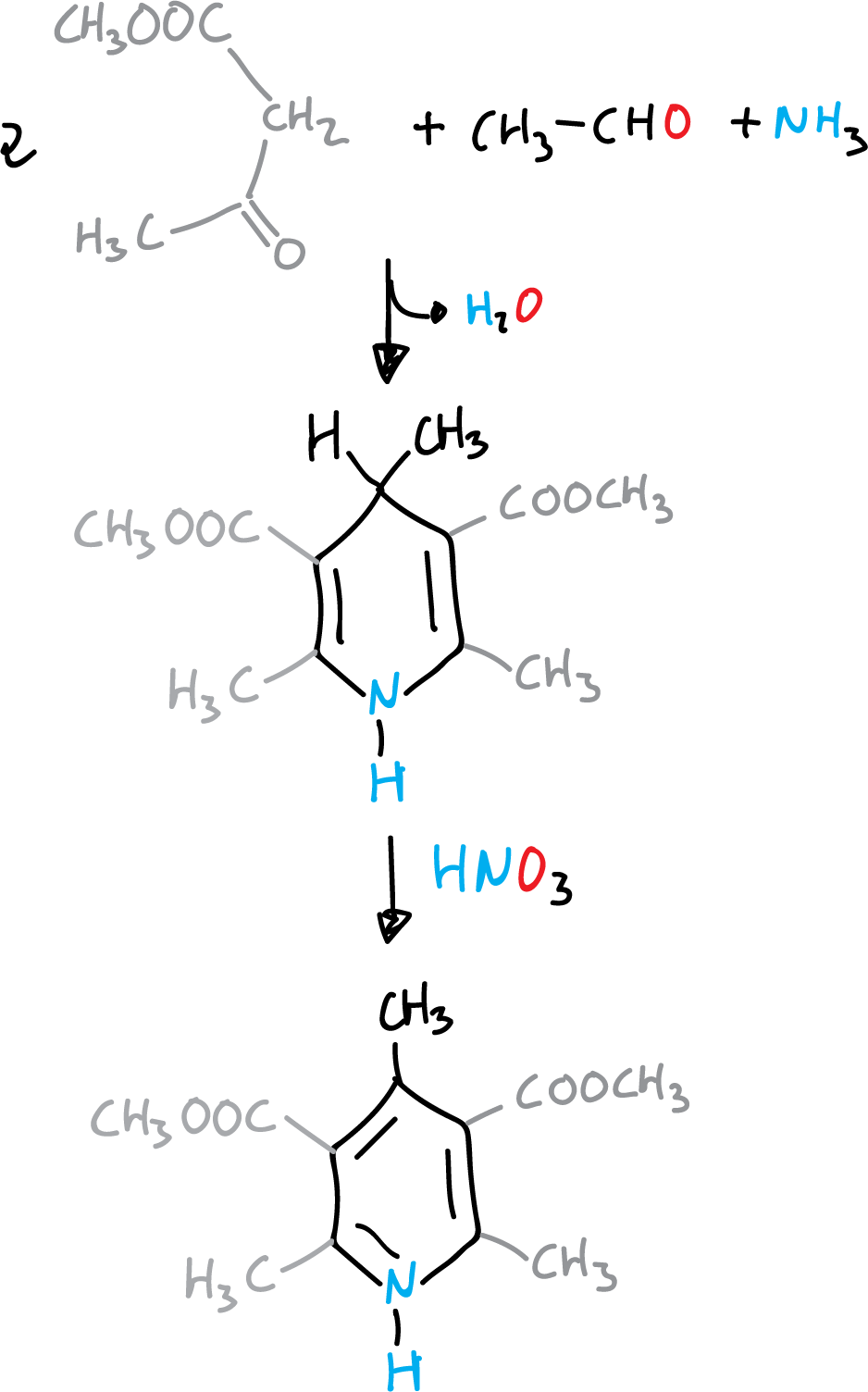
Mecanismo de reacción
La primera etapa es una condensación de Knoevenagel del β-cetoéster y el aldehído.
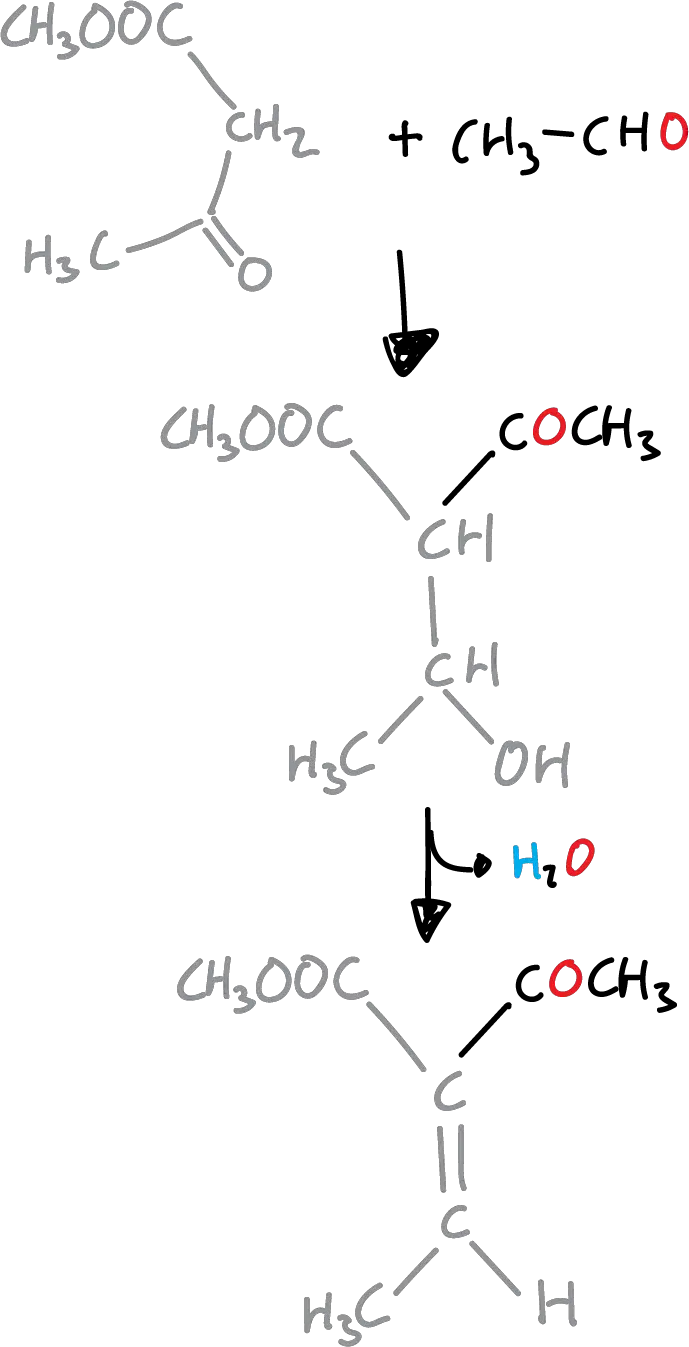
A continuación, el cetoéster insaturado que se forma sufre una condensación con la enamina producida con el amoniaco y el β-cetoéster original, para dar la dihidropiridina correspondiente.
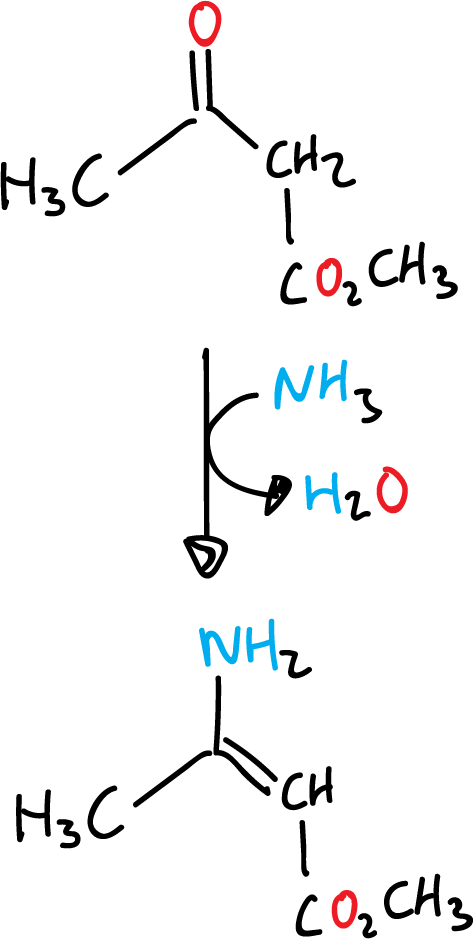
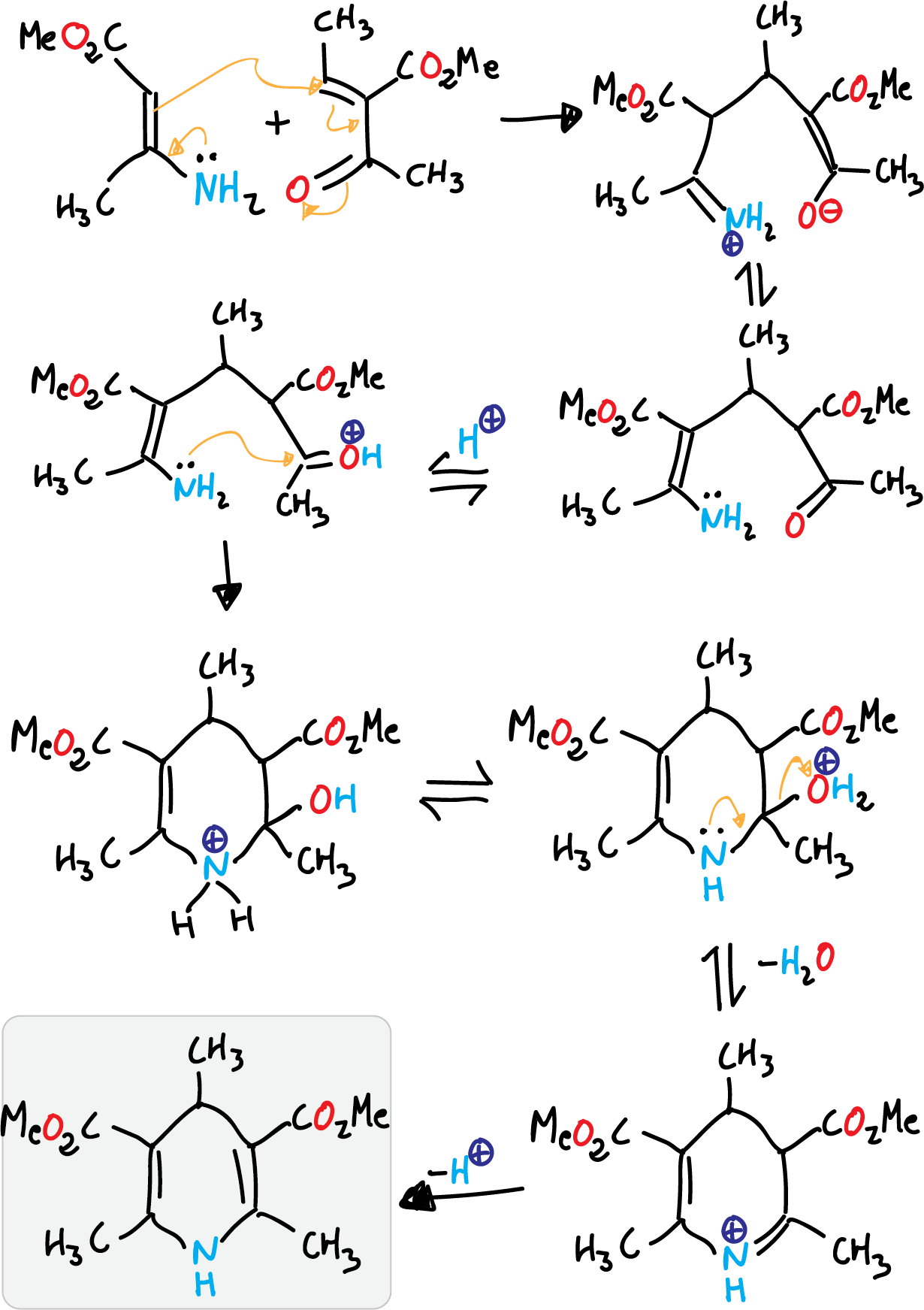
Esta reacción corresponde a la ruta A del análisis retrosintético anteriormente realizado.
Otras variantes
Se han descrito muchas modificaciones y variantes de la síntesis de Hantzsch de piridinas.
Por ejemplo, se puede emplear acetato de amonio (AcONH4) con ácido acético (AcOH), en lugar de amoniaco. Este método sigue la ruta c, del anterior análisis retrosintético. Así, en el primer paso se forma enamina, que en la siguiente figura se indica como I que reacciona con otra molécula del reactivo.
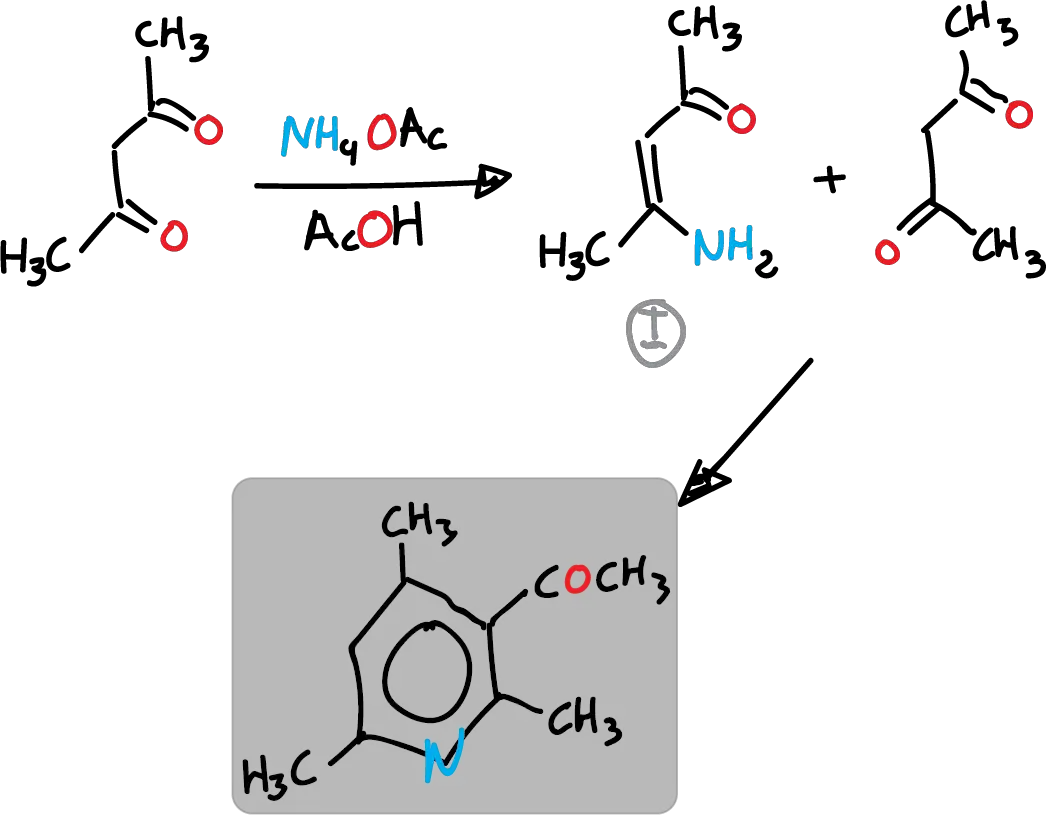
La ruta B, que es la ciclación de 1,5 dicetonas con amoniaco, tiene aplicación más bien limitada. Sin embargo, se describe a continuación una variante más versatil denominada síntesis de Krönke.
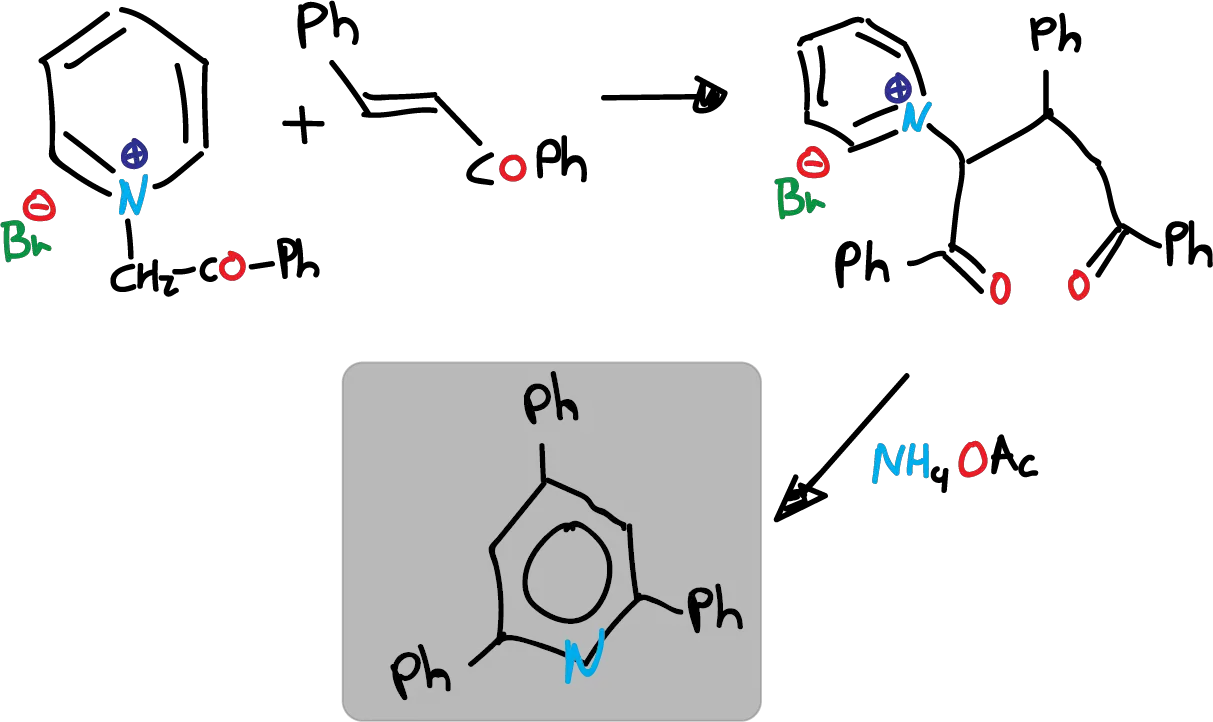
Se trata de la adición de un iluro de piridinio a un compuesto carbonílico α,β-insaturado, que lleva a la formación de una 1,5-dicetona. Esta 1,5-dicetona, se encuentra en el estado de oxidación apropiado para ciclarse directamente.
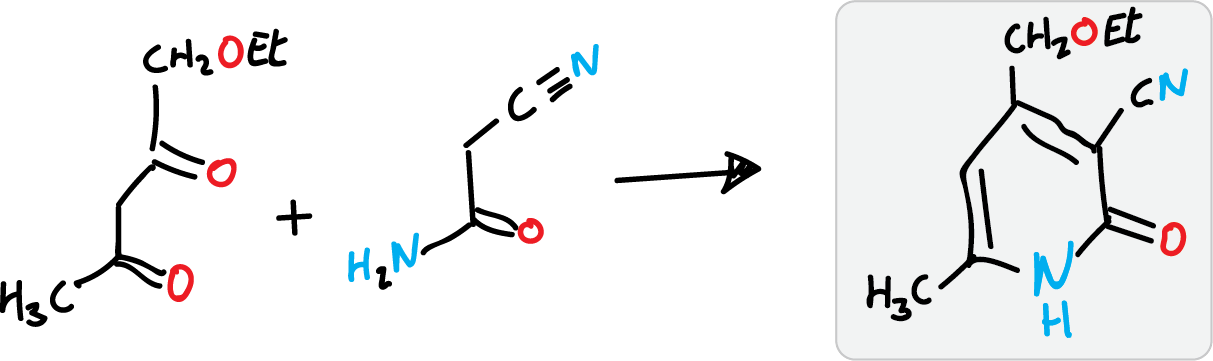
Síntesis de Guareschi-Thorpe de 2-piridonas
Se utiliza para preparar 2-piridonas, mediante la cianoacetamida como componente nitrogenado y con una 1,3-dicetona o bien un β-cetoéster.
Otras variantes
Otros métodos de síntesis se han descrito.
- Por ejemplo, partiendo de amoniaco y unidades de 5 átomos de carbono (como el aldehído glutacónico).
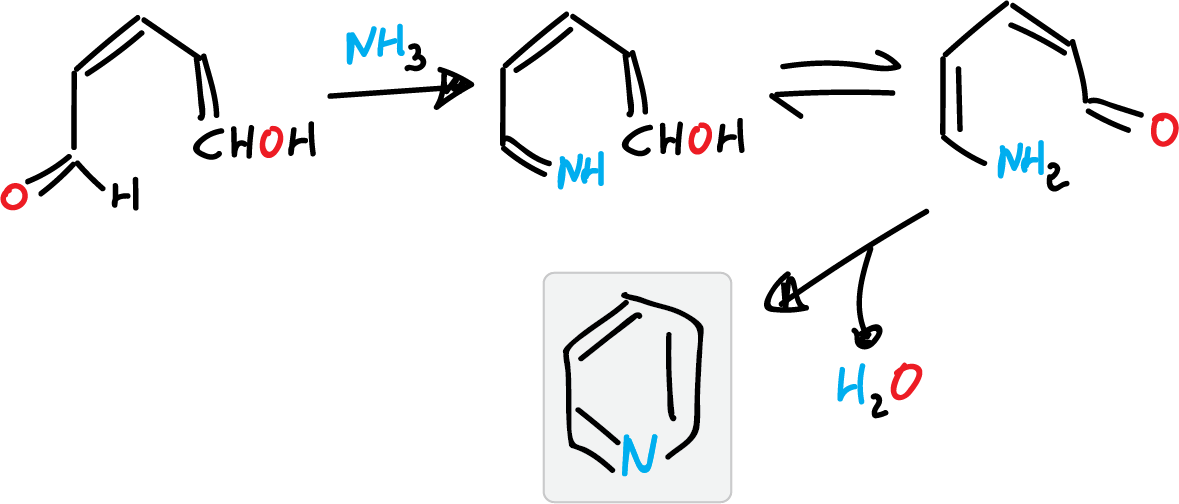
Esta reacción no tiene aplicación práctica ya que el aldehído glutácónico se obtiene por la apertura de un anillo de sal de piridinio.
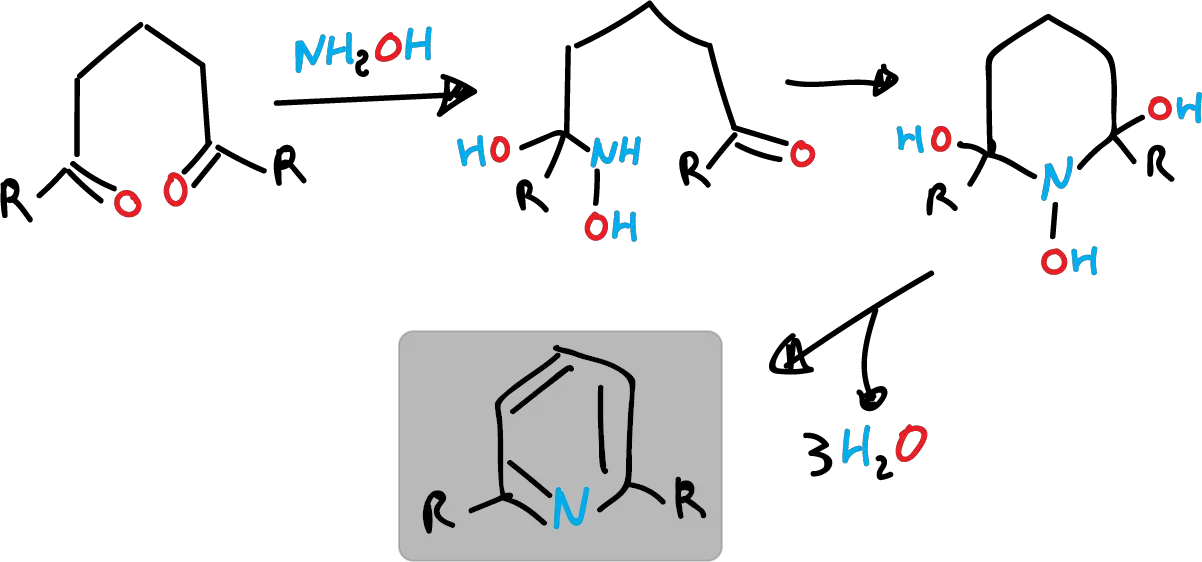
- De forma análoga, reaccionan los compuestos 1,5-dicarbonílicos con la hidroxilamina (NH2OH).
Síntesis de piridinas sustituidas
La síntesis de las piridinas sustituidas, en lugar de realizarse a partir de la piridina, se obtienen a partir de los distintos compuestos alifaticos que se han descrito anteriormente, convenientemente sustituidos. Esto es debido a que la sustitución electrofílica aromática (SEAr), no está favorecida en el anillo piridínico. Pues los electrófilos atacan con frecuencia al nitrógeno básico.
No obstante, hay una gran variedad de reacciones que dan la piridina y derivados.
Propiedades químicas de piridinas
La piridina es el compuesto heterocíclico más parecido al benceno. Presenta una alta energía de resonancia y su estructura se parece mucha a la del benceno. Naturalmente, la presencia del nitrógeno le confiere unas características especiales. El par de electrones en el plano del anillo, ofrece un centro susceptible de protonación y alquilación.
Muchas de las propiedades químicas de la piridina serán, por tanto, la de una amina terciaria y el sexteto aromático no interviene en estas reacciones.
Como se ha mencionado, la reacción característica del benceno, la sustitución electrofílica aromática (SEAr), no es evidente en las piridinas.
En primer lugar el producto de la reacción de una piridina con un electrófilo es aquel en que éste se halla coordinado con el par de electrones del nitrógeno. Por tanto, dificulta la sustitución en los átomos de carbono del anillo, o sea, que se favorece la formación inicial de una sal de piridinio.
Por ejemplo, la acción del pentóxido de dinitrógeno (N2O5) sobre piridina, en condiciones neutras da:
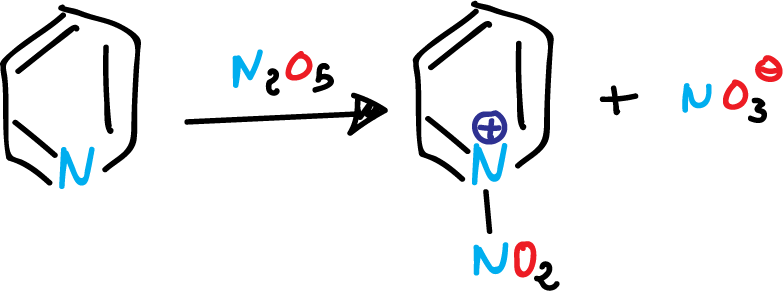
No obstante, cuando las posiciones C2 y C6 del anillo de piridina se encuentran sustituidas por grupos voluminosos, se impide estéricamente la coordinación el el nitrógeno. Por tanto, la sustitución en el anillo se puede producir en condiciones relativamente suaves.
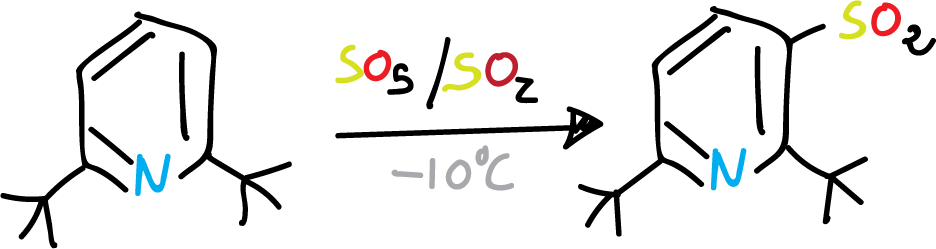
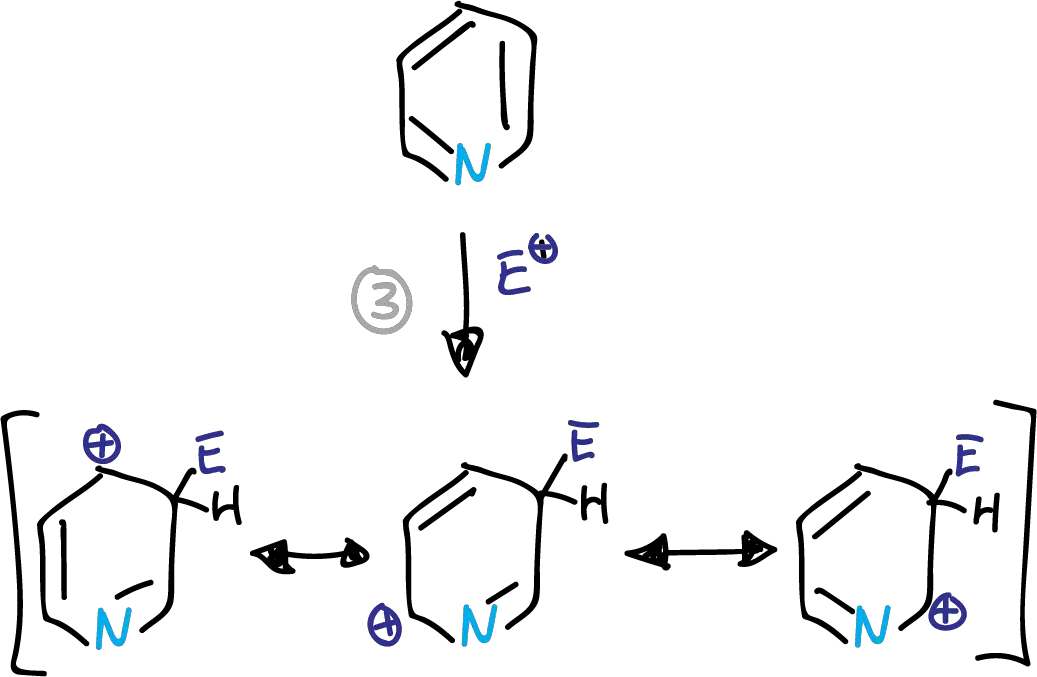
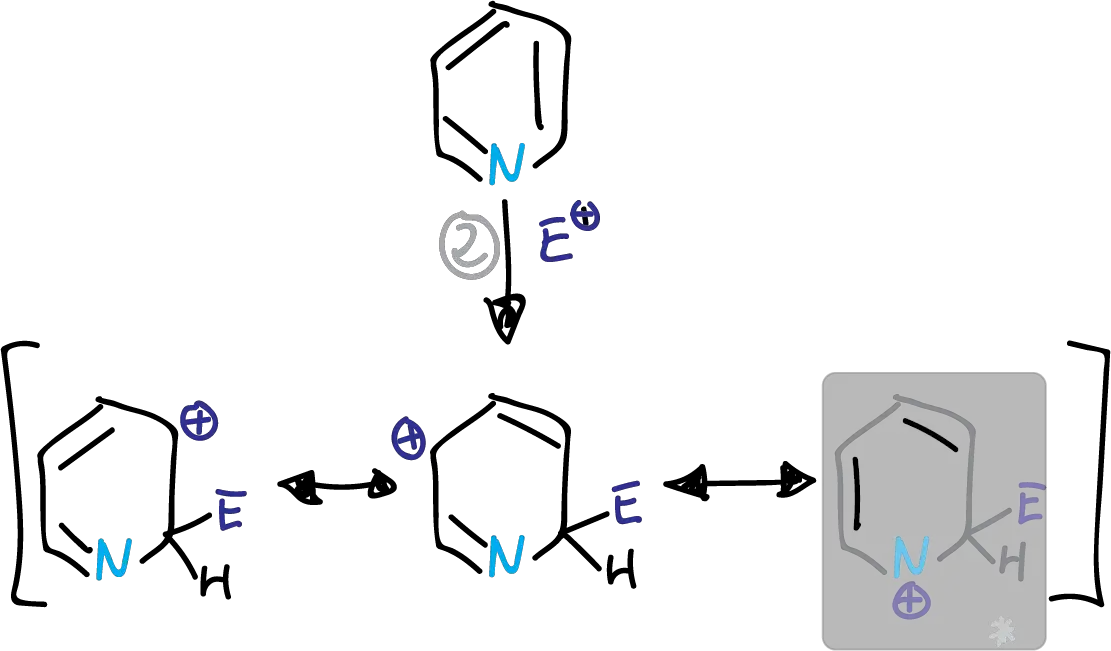
la SEAr en la piridina e iones piridinio, se produce preferentemente en la posición C3. Esto es debido a los correspondientes intermedios en el estado de transición, como se indica en el esquema.
Tanto si la sustitución se produce en C2 o en C4, nos encontramos con una forma desfavorecida que está resaltada en las figuras.
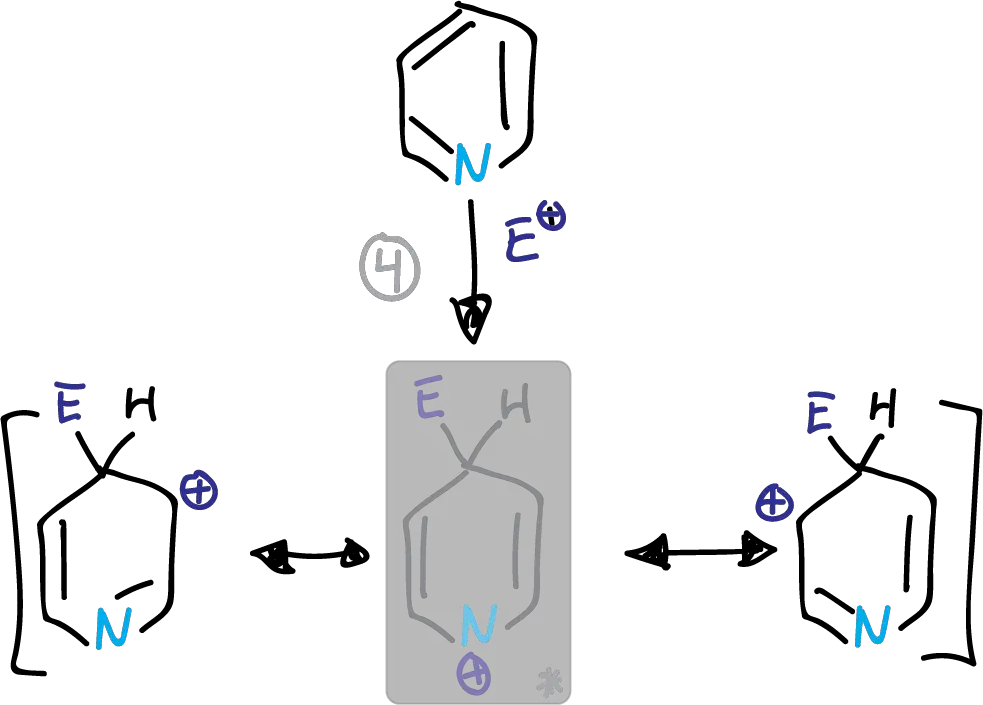
Esta estructura presenta una carga positiva parcial sobre el nitrógeno electronegativo. Lo cual provoca que aumente la energía del estado de transición. Por tanto, la sustitución se realiza en la posición C3, preferentemente.
Se ha observado que las sustituciones alquilo activan la piridina, favoreciendo la sustitución elecrofílica. Por ejemplo en la nitración.
Para nitrar la piridina se requieren condiciones vigorosas (H2SO4—SO3 /NO3K, 300 ºC) para dar 3-nitro piridina, con bajo rendimiento.
Con un sólo grupo metilo no es suficiente para activar la piridina, y mas bien la mezcla nitrante la oxida. Sin embargo, si introducimos varios metilos, la reacción se da con mayor facilidad.
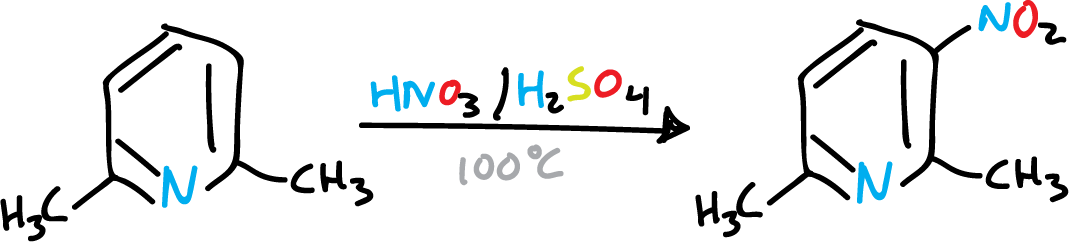
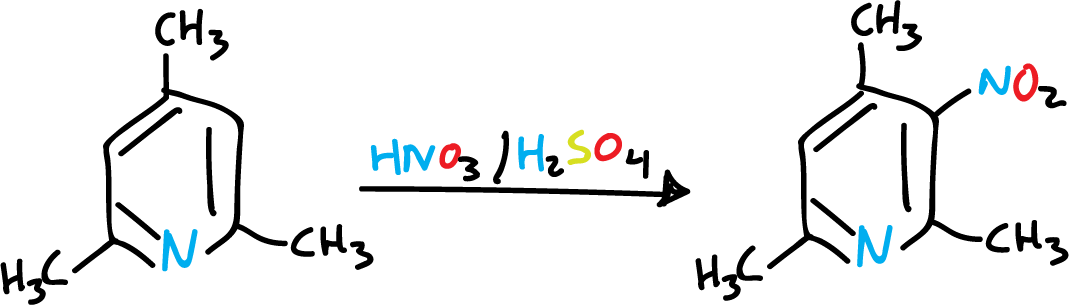
Sin embargo, de una forma general, el átomo de nitrógeno del heterociclo sigue siendo la influencia orientadora que predomina en las piridinas, como se puede ver en la siguiente sulfonación.
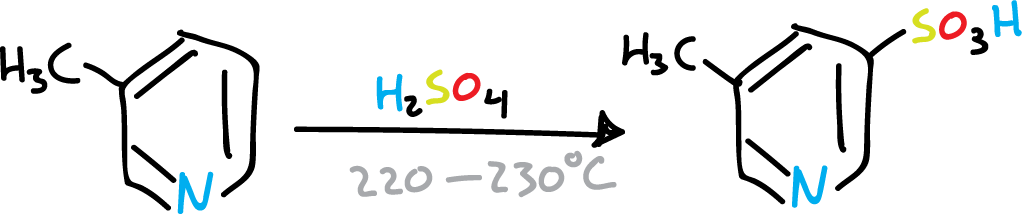
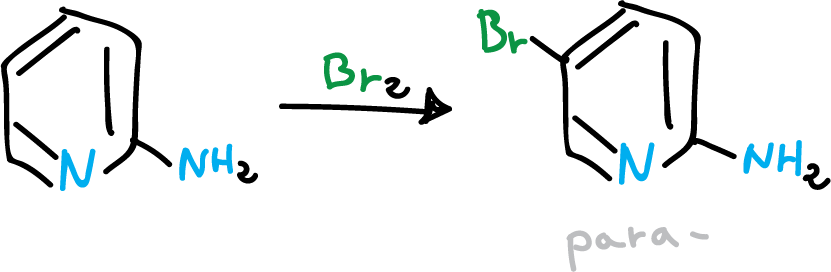
La sustitución posterior de piridinas halogenadas, también está controlada por el centro heterocíclico.
Cuando el sustituyente de la piridina es un grupo amino, estos son los que ejercen el efecto orientador dominante, facilitando la sustitución.
Así, un grupo amino en C2 dirige a C5 (para– dirigente).
Se el grupo amino está en C3 orienta a C2 y si se encuentra en C4 dirige a C3 (orto– dirigente).

Tambien, predominan en a orientación los grupos hidroxilo en C3.
fig-22
Si el hidroxilo está en C2 o C4, estas estructuras existen predominantemente en forma de piridona, y como es de esperar esto influye en su reactividad.
Por ejemplo, las 2-piridonas experimentan un ataque en posición C3, preferentemente, lo cual contrasta de forma notable con el comportamiento de los derivados 2-alcóxido, en los que se sustituye en C5.
fig-23
Los N-óxidos de piridina también puden sufrir el ataque electrofilico en la posición C3 y C4 dependiendo de que la reacción se efectúe con una base libre o con el ácido conjugado. La nitración se efectúa mediante ataque sobre la base libre.
fig-24
De esta forma, se lleva a cabo en la posición C4. Cuando esta posición está ocupada, la nitración no se produce.
La sulfonación se produce con dificultad en la posición C3.
fig-25
Cuando hay grupos alquilo se activan, al igual que en el caso de la piridina y aquí también predomina el efecto orientador del heterociclo.
En cambio, con sustituyentes amino el orden del poder direccional parece ser: NR2 > N⊕—O⊖ > NHCOR.
fig-26
Los grupos N-óxido tienen mayor fuerza directora comparada con los sustituyentes alcóxido, en los productos oxidados.
fig-27
Sin embargo, los grupos hidroxilo en C3 presentan bastante fuerza directora (comparado con N-óxido) y su influencia predomina, como en el caso de la piridina.
fig-27
El N-óxido de 2-hidroxi-piridina (N-hidroxi-2-piridona) experimenta bromación para dar el derivado 3-bromado y al someterse a la nitración, se observa una sustitución en la posición C5.
fig-28
Las 4-piridonas y las N-hidroxi-4-piridonas se comportan como era de esperar y suelen experimentar sustitucion en C3 y C5.
Es evidente que las piridinas y sus N-óxidos carezcan de la reactividad necesaria para interactuar con los electrófilos más débiles del tipo generado en las reacciones Friedel-Crafts y Vilsmeier–Haack (formilación con Ph(Me)NCHO y POCl3).
Video de las Piridinas
Sustitución nucleófila de piridinas
Reacciones en sustituyentes de piridinas
Catedrático de Química Orgánica en la Universidad de Granada, con una larga trayectoria en Química Computacional, en modelado y diseño molecular.