Índice
Bencenos sustituidos
Como podemos observar no es muy amplia la variedad de sustituyentes que se pueden introducir directamente sobre el anillo aromático. Sin embargo, es posible la transformación posterior de éstos en otra gran variedad de funciones mediante reacciones relativamente simples, así por ejemplo:
Halogenación de alquilbencenos en posición bencílica
Los hidrógenos bencílicos presentan una reactividad especial. Es por ello, que una de esas reacciones es la sustitución por halógenos mediante un mecanismo radicalario. Con todo esto, sólo tienen aplicación sintética la cloración y la bromación. Así, se pueden utilizar Cl2 o Br2 calentando o bien iluminado con una radiación de longitud de onda adecuada. Alternativamente, se pueden utilizar como fuentes de halógeno N-clorosuccinimida (NCS) o N-bromosuccinimida (NBS).
En estas condiciones, no se produce reacción alguna sobre el anillo aromático. Por consiguiente, la clave del éxito de esta reacción recae en la estabilidad del grupo bencilo (intermedio de la reacción).
Por otro lado, los haluros de bencilo son compuestos útiles usados en reacciones SN2 o en la preparación de alquenilbencenos.
fig1
fig2
Oxidación de alquilbencenos
Los derivados del benceno con una cadena alquílica con al menos un hidrógeno en la posición bencílica pueden oxidarse para dar un grupo carboxilo, independientemente de la longitud o ramificación del grupo alquilo. Como oxidante se suele emplear K2Cr2O7 o K2MnO4 en medio ácido y en caliente.
fig3
Reducción de aldehídos y cetonas aromáticas (Clemmensen), (Wolff-Kishner)
Las cetonas aromáticas se pueden reducir hasta el correspondiente compuesto saturado por dos procedimientos:
1) Reducción de Clemmensen: tratamiento con amalgama de zinc y HCl en caliente.
2) Reducción de Wolff-Kishner: tratamiento con hidrazina en medio básico en un disolvente de alto punto de ebullición.
fig4
En estas condiciones no se reducen ni los dobles enlaces, C=C, ni los triples enlaces, C≡C, ni el grupo COOH. La elección de un método u otro va a depender de la compatibilidad del resto de las funciones con las condiciones ácidas de la reacción deClemmensen o básicas de la Wolff-Kishner.
Estas reacciones, junto con la acilación Friedel-Crafts, son una alternativa a la alquilación Friedel-Crafts cuando se trata de introducir una cadena alquílica a partir de un haluro de alquilo primario, ya que como se ha visto con anterioridad, se producen transposiciones cuando se trata de introducir cadenas alifáticas largas por alquilación directa, véase alquilación Friedel-Crafts.
fig5
Oxidación de alcoholes bencílicos
Los grupos hidroxilo en posición bencílica se oxidan hasta grupos carbonilo con oxidantes suaves como MnO2 (dióxido de manganeso).
fig6
Reducción de grupos nitro
Los grupos nitro unidos a anillos de benceno pueden reducirse fácilmente hasta amina por varios procedimientos. Los más empleados en nitrocompuestos aromáticos son:
- Hidrogenación catalítica en presencia de Pt como catalizador.
- Tratamiento con metales (Fe o Sn) y ácido clorhídrico. Con ácido clorhídrico, se obtiene el clorohidrato de la amina correspondiente.
- Tratamiento con SnCl2 y ácido clorhídrico.
fig7
Hidrólisis de grupos -SO3H
La reacción de sulfonación en el benceno es reversible, por lo que calentando el ácido sulfónico con una disolución acuosa de H2SO4 o H3PO4 es posible obtener de nuevo el benceno. Tiene utilidad sintética para dirigir otras reacciones de sustitución aromática electrofílica hacia las posiciones que activen el grupo SO3H y eliminarlo después.
fig8
Conversión de haloalcanos en compuestos organometálicos
Los haluros de arilo tratados con metales como Li o Mg dan compuestos organometálicos, análogamente a como reaccionan los haluros de alquilo. Dichos compuestos reaccionan de forma similar a los organometálicos alifáticos.
fig9
Arilaminas
Sustitución aromática electrofílica de arilaminas
Son sustancias muy reactivas en la sustitución aromática electrofílica, ya que los grupos -NH2, -NHR y -NR1R2 son fuertemente activantes y orto- y para- dirigentes, y suelen darse productos de polisustitución. Para la anilina es posible realizar la yodación directa tratando con I2 y en presencia de una solución de bisulfito sódico, NaHSO4. Con Br2 o Cl2 se pueden dar productos de polisustitución.
Las reacciones de nitración, sulfonación alquilación o acilación no se pueden realizar de forma directa ya que, o bien reacciona con el ácido que se emplea como catalizador o se oxidan o polimerizan como con los ácidos nítrico y sulfúrico, respectivamente. Para evitar estos problemas, se convierte el grupo -NH2 en una amida mediante una reacción de acilación.
fig9
Acilación de arilaminas
El grupo -NH2 se puede convertir en una amida por tratamiento con un cloruro de acilo o anhídrido. No es necesario añadir una base, ya que la propia basicidad del grupo -NH2 cataliza la reacción. Generalmente, se emplea cloruro de acetilo o anhídrido acético. Se regenera la amina por tratamiento con una disolución de NaOH en caliente o con ácido clorhídrico en caliente. En este caso se aísla el clorohidrato de la amina.
Otra característica del grupo que se genera es su carácter activante, aunque menor que el del amino libre, debido a que se retira densidad de carga del anillo de benceno.
fig9
Síntesis y reactividad de las sales de diazonio
Las sales de diazonio en compuestos aromáticos presentan un elevado interés en síntesis orgánica, ya que a partir de ellas es posible preparar una gran variedad de derivados.
Se obtienen por tratamiento de una amina con nitrito sódico en medio ácido a baja temperatura, ya que se descomponen perdiendo nitrógeno con relativa facilidad, si se eleva la temperatura.
Las sales de diazonio pueden sufrir las siguientes reacciones:
1. Reducción con ácido hipofosforoso para dar el correspondiente areno.
2. Hidrólisis catalizada por ácidos para dar fenoles.
3. Reacciones de sustitución:
- Obtención de fluoruros de arilo por reacción de Schiemann:
- Reacciones de tipo Sandmmeyer: son un conjunto de reacciones donde la sal de diazonio se desplaza por un nucleófilo como Cl⊖, Br⊖, I⊖, CN⊖ o R-SH, respectivamente. Los grupos -CN son precursores de ácidos carboxílicos (-COOH) ya que la hidrólisis en medio ácido se produce la transformación -CN → -COOH. En muchos casos, se requiere la catálisis con Cu(I), por lo que se emplean las sales de cobre de los aniones. Para los tioles o el yoduro, no es necesario la presencia del catalizador. Esta reacción es importante ya que se obtienen productos que por sustitución aromática electrofílica directa del correspondiente areno no pueden realizarse.
- Una alternativa para la preparación de arilnitrilos parte de haluros de arilo, que con CuCN dan el producto deseado (reacción de Rosenmund-von Braum).
4. Azocopulación: la reacción de sales de diazonio con arenos activados en medio neutro o ligeramente ácido, conduce a la formación de azo-compuestos (-N=N- grupo azo). En anillos de benceno la sustitución se produce, preferentemente, sobre las posiciones para- del grupo activante. Es un método empleado ampliamente en la síntesis de colorantes.
Fenoles
Reacciones ácido-base de fenoles
Los fenoles reaccionan con bases para dar fenóxidos, que son unos excelentes nucleófilos.
fig12
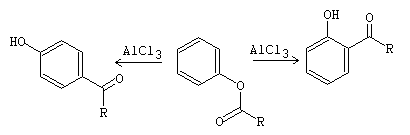
Reacciones de acilación de fenoles y transposición de Fries
La acilación del –OH se da en medio básico y la transposición, en medio ácido. Así, se pueden obtener ésteres de fenol empleando anhídridos de ácido y AlCl3, en presencia de una base como la piridina. Generalmente, se utiliza anhídrido acético.
fig13
fig14
Los grupos acilo amortiguan el carácter fuertemente activante del grupo –OH del fenol, debido que se atenúa el efecto electrón donante del grupo hidroxilo. Por tanto, esto es debido a que producen una disminución de la densidad de carga en el anillo aromático.
Así, la reacción de C-acilación de fenoles se da en condiciones de control termodinámico. Debido a que el producto obtenido es más estable. Sin embargo, el producto de O-acilación (esterificación) se da en condiciones de control cinético.
Por otro lado, los ésteres de arilo, resultantes de la reacción de O-acilación, se transforman, con AlCl3, en las correspondientes arilcetonas. A esta reacción se la conoce como transposición de Fries.[1]
fig15
Reacciones de oxidación de fenoles
Los fenoles se oxidan más fácilmente que los alcoholes. Son muchos los reactivos que se pueden emplear, destacando Ag2O o Na2Cr2O7. Además, son especialmente de relevancia, sobre todo en sistemas redox biológicos, las oxidaciones de 1,2- y 1,4-difenoles que generan benzoquinonas.
fig16
Reacciones de sustitución aromática electrofílica de fenoles
Los fenoles son muy buenos sustratos para la reacción de sustitución aromática electrofílica. Y como se ha comentado anteriormente, el grupo OH es fuertemente activante y orto- y para- dirigente.
Por tanto, debido a la mayor reactividad de los fenoles, se suelen obtener productos de polisustitución. Así, las condiciones de las reacciones de sustitución aromática electrofílica son más suaves que las de la reacción de sustitución del benceno.
Carboxilación de fenoles (reacción de Kolbe-Schmitt)
La reacción de Kolbe-Schmitt es una reacción de transformación de fenoles, promovida por bases, que lleva a la formación de ácido salicílico y derivados.
fig17
Resumen
Las diferentes reacciones de sustitución aromática electrofílica vista en los apartados anteriores para compuestos aromáticos sustituidos se pueden resumir en la siguiente tabla para derivados de benceno, fenol y anilina.
| Reacción | Benceno | Fenol | Anilina | |
| Halogenación | X2 / FeX3 | X2 sin catalizador | X2 sin catalizador < (I2 / HCO32-) | |
| Productos de polisustitución | ||||
| Nitración | HNO3 / H2SO4 | HNO3 / H2O | — | |
| Rendimiento bajo por oxidaciones, mezcla de orto- y para- | — | |||
| Sulfonación | H2SO4 fumante | H2SO4 concentrado | — | |
| Mayoritario orto- (control cinético) | — | |||
| Alquilación | R-Cl / AlCl3 | R-OH en medio ácido o R-Cl / AlCl3 | — | |
| Acilación | RCOCl / AlCl3 | RCOCl / AlCl3 | CH3COCl | |
Referencias y notas
[1] a) K. Fries, G. Fink, Chem. Ber. 41, 4271 (1908). b) K. Fries, W. Pfaffendorf, Chem. Ber. 43, 212 (1910).
Volver a la página de Síntesis y Reactividad de Compuestos Orgánicos.
Catedrático de Química Orgánica en la Universidad de Granada, con una larga trayectoria en Química Computacional, en modelado y diseño molecular.